Metastasis
By Linda Meade-Tollin, Fred Bosman
The spread of cancer cells to distant sites implies a complex series of cellular abnormalities caused, in part, by genetic aberrations
The spread of cancer cells to distant sites implies a complex series of cellular abnormalities caused, in part, by genetic aberrations
DOI: 10.1511/1998.21.130
Mary is a 46-year-old mother of two daughters, ages 17 and 19. Eight years ago she was diagnosed with breast cancer, and after a conventional course of chemotherapy followed by radiation, the cancer was driven into remission. Lately she is having increasingly severe back pains.
At a routine check-up, she mentions the pains to her surgeon, who performs a bone scan. The scan reveals several "hot spots," regions of increased metabolic activity in her spinal column. The diagnosis made on a biopsy of one of these hot spots catches her completely off guard: metastasis of her breast cancer.
Patients like Mary inevitably ask the question: Why me? But for those of us who study what cancer is, its development and spread, the question is rather: Why at all?
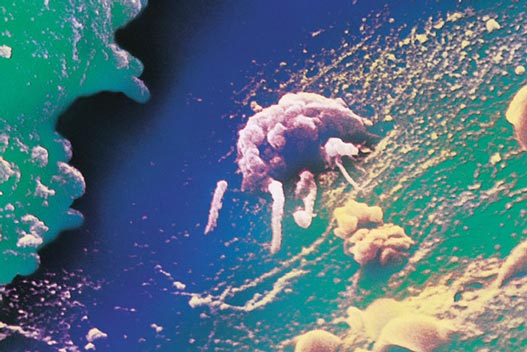
The simple answer is that in cancer the genes and chromosomes of cells become disorganized, leading them to enact genetic programs far different from the intended normal program. It has often been noted that cancer cells are unusual because they are undifferentiated; that is, they lose functions and, as a result, fail to fully develop the characteristics and proper activities of mature cells of their type. Quite remarkably, metastatic cancer cells gain new functions, taking on characteristics unrelated to the normal, often sedentary cell type.
Most cells are designed to remain fixed in their organs in order to perform the specialized activities for which they are particularly adapted. They are held tightly in place by molecular tethers that link them with the other cells and the proteinaceous matrix in that tissue. Clearly, these cells are not meant to roam around the body and take up residence in other organs. Metastatic cancer cells are altered in such a way that they can break those bonds. Further cellular remodeling allows metastatic cells to chew through the proteinaceous walls that line tissues and blood vessels and quite literally walk away from their parent organ to invade other tissues and organs.
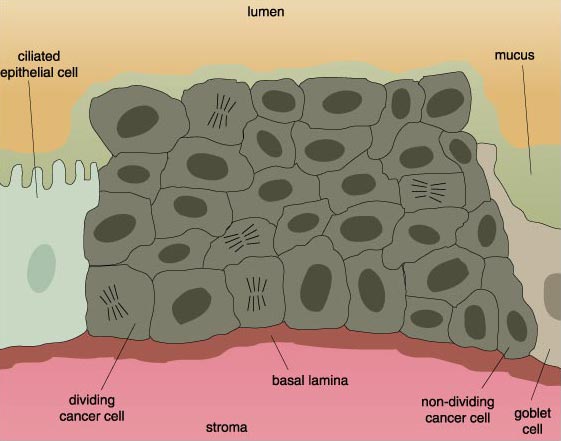
If it were not so potentially damaging, metastasis might be just an interesting cellular oddity. But the fact is that the metastatic tumor is often more dangerous than the original, or primary, tumor. Metastatic cancer cells crowd out normal cells in the tissue and deprive them of nutrients. In effect, the metastatic cells starve and displace functional cells. This can be lethal when the organ—or organs, since cancer cells can go almost anywhere and often end up in several new organs—can no longer perform its vital functions.
Over the past few decades, biologists have become ever more precise in determining the genetic, biochemical and cellular changes that drive a cell first to become cancerous and then to become metastatic. And new discoveries are being reported daily. So, although not all of the details are entirely known, a comprehensive picture of these alterations is emerging. Each new piece of information not only contributes to this overall picture; it also provides a site of potential therapeutic intervention to inhibit the progression of cancer—if not, someday, to eliminate it entirely.
A diagnosis of cancer marks an abrupt change in the life of the patient. A line of demarcation has been drawn. Life seems to be separated into before and after. Yet the events that lead a cell to become cancerous take place gradually, sometimes over periods that can exceed 10 years. During this lengthy evolution, cells undergoing cancerous transformation accumulate genetic abnormalities, one important consequence of which is that cellular growth becomes deregulated.
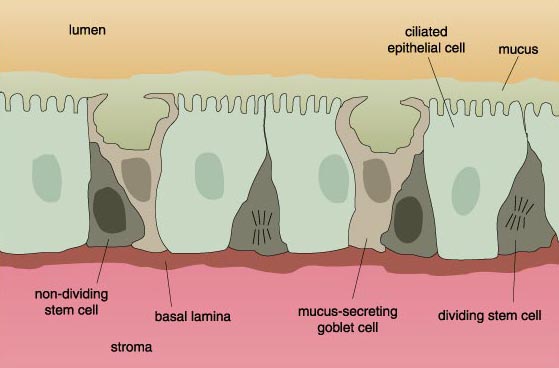
The number of cells in normal tissue is strictly controlled by a system of checks and balances. Cells that are too old, that fail to function properly or that are otherwise no longer needed are programmed to die, a process known as apoptosis. These cells are replaced by new cells, derived from primitive precursors, also called stem cells, that divide and then differentiate into the mature cell type that performs a specific function. In time, these too undergo apoptosis, only to be replaced by younger cells.
Cell death does not take place because a cell just falls apart. Rather it is the result of a carefully controlled genetic program. Given the proper signals, particular genes are activated that encode proteins that carry out the cellular suicide.
Not all cells undergo apoptosis. There is, in fact, a striking variation in the longevity of different cell types. Nerve and muscle cells, for example, are extremely long-lived—if all goes well, they endure for the entire life of the individual. Once these cells reach their fully mature state they can no longer divide, and it is unlikely that there are populations of stem cells that can generate replacements when they die.
Other cells are actually designed to die shortly after reaching maturity. Often these are the cells that turn over frequently, such as skin cells, or the epithelial lining of the digestive system. The epithelial cells lining the digestive tract mucosa live for a maximum of four days after reaching maturity. High-turnover tissues are endowed with a population of stem cells ready to replenish their respective tissues with mature cells, as older cells are sloughed off, used up or eliminated. For example, stem cells in the bone marrow divide continuously to provide sufficient numbers of blood cells. Similarly, the stem cells in the digestive tract are constantly dividing to replace worn-out mucosal epithelial cells.
Some tissues maintain cell-growth rates intermediate between these two extreme scenarios. For example, under normal circumstances, cell division in the liver takes place at a very low rate: At any given time, only one in 10,000 liver cells is dividing. But cell-division rates can be driven up when needed, for instance, to replace cells damaged by viral infection or removed by surgery.
Clearly, then, maintaining the proper number of cells in any tissue requires a delicate balance between cellular production and elimination. Essential to this balance is apoptotic cell loss on one hand and cell division on the other. When an excess of cells is produced by unrestrained cell division, or when too few cells are eliminated by apoptosis, an overabundance of cells accumulates in the tissue. This basically describes the situation in cancer.
As with all cellular functions, a cell's life and death are under strict genetic control. These genes encode proteins that sense growth signals from the environment, or drive the cell through its replicative cycle and check for cellular and genetic abnormalities. Some of these regulatory proteins make the necessary corrections to damaged genes. Other proteins direct the cell to exit the cell cycle in order to differentiate. Still other proteins remove old or defective cells from the replicative cycle and send them en route to apoptosis.
It stands to reason that if any of these genes or the proteins they encode become faulty, the cell-division cycle will become abnormal. For example, alterations to the series of proteins that detects external growth cues can lead to an abnormally prolonged growth cue. Problems with the proteins that regulate the cell cycle can, likewise, maintain cells in a proliferative mode when they normally would exit the cycle and start to differentiate and mature.
In recent years, scientists have found that in the majority of cancer cells, one or several of these growth-regulatory genes is either missing or defective. It should be noted that all of these genes, when functioning properly, are crucial in maintaining the normal growth characteristics for each cell type. These genes only promote cancer when they become mutated or altered.
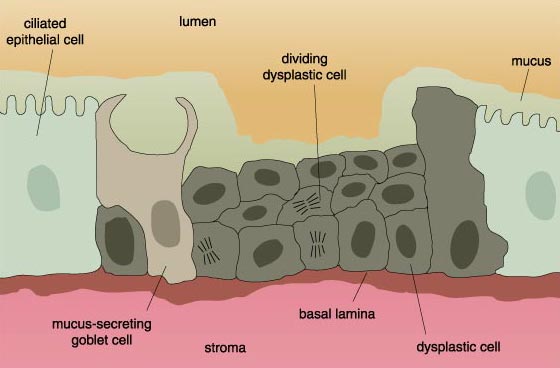
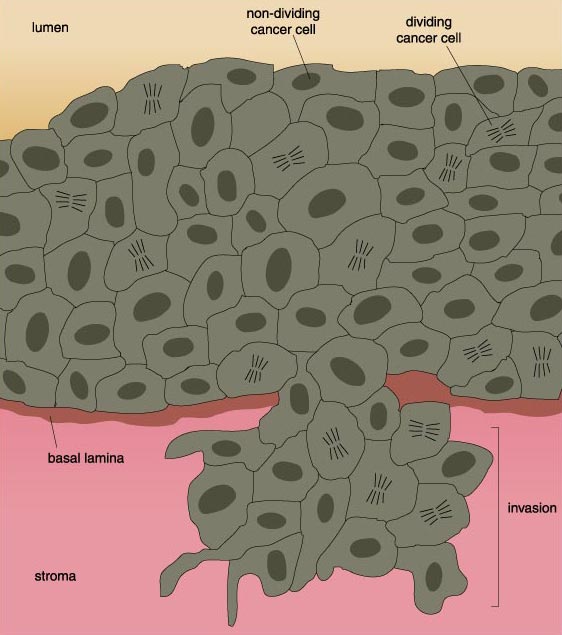
Cells on their way to becoming cancerous accumulate a number of genetic and chromosomal abnormalities, each of which in some way pushes the cell further in the direction of unrestricted growth. At first, clusters of genetically identical cells are formed, each cell dividing with less restraint than its normal neighbors. This cluster does not at this point constitute a tumor.
When the cell mass attains a diameter of about 2 millimeters, the cells emit signals that recruit surrounding connective tissue and vascular cells to the tumor and induce them to grow into blood vessels. The cell mass literally stimulates the growth of its own blood supply from existing blood vessels, a process called angiogenesis. There is some evidence that angiogenesis is probably initiated because cells in the mass, especially those in the interior, become starved for oxygen. In any event, once the blood supply is in place, the cell mass can import oxygen and the nutrients it needs to keep growing. Furthermore, the cells now have a conduit through which they can escape and invade other tissues. Angiogenesis seems to have an additional consequence. Connective tissue surrounding blood vessels release factors (as do the vascular cells) that stimulate the growth and motility of cancer cells. Strange as this may sound, cancer can only develop if the cancer cells are adequately supported by host tissues.
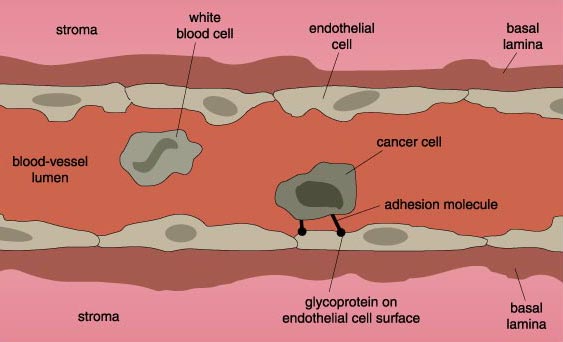
The escape of cancer cells is not a trivial matter. As mentioned above, most normal cells are tightly hooked in place by chemical bonds linking them to neighboring cells, to the surrounding connective tissue matrix—the mortar of tissues—or both. This is especially true of the epithelial cells that line many organs and from which the majority of cancers arise. The cells are held in place by large molecules on their surfaces that connect with similar proteins on neighboring cells or with proteins in the matrix. In addition to these molecular tethers, most normal cells simply lack the migratory machinery of mobile cells.
There are, of course, some important exceptions, most notably the white blood cells that constitute the immune system. These are designed to migrate out of the blood stream and into tissues to fight infection. In order for sedentary cells to metastasize, they must lose their adhesive connections and take on the migratory properties usually restricted to white-blood cells.
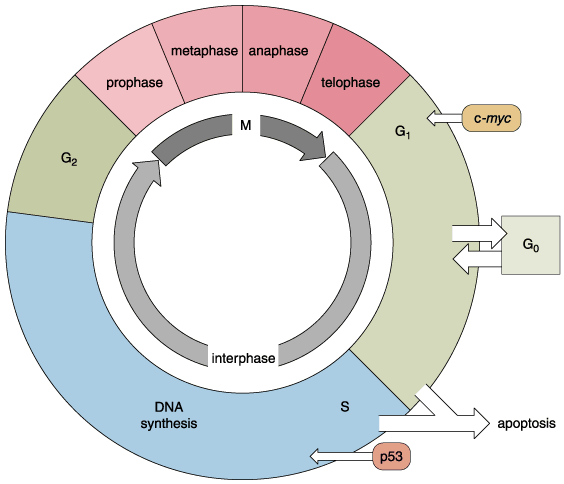
Finally, the cell undergoes Mitosis (M), during which each daughter cell receives a complete set of the cell’s genes, along with all of the other constituents it needs to function and mature. To ensure that cell division equals apoptotic cell loss the cell’s response to growth cues is regulated, for example, by the protein Myc, encoded by the gene c-myc, which exerts its effects during the G1 phase of the cycle. In order that DNA be faithfully duplicated, checkpoints exist at certain steps of the cell cycle. The p53 protein mediates a checkpoint when the cell enters the S phase. If all is well, the cell continues with the cycle. But if the DNA is damaged, the cell cycle is stopped to allow for DNA repair. If the damage is so severe that it cannot be fixed, the cell exits the cycle and instead undergoes apoptosis. Mutations in the c-myc gene, in the p53 gene or in any of the many other regulatory genes, or the genes that encode proteins that correct defects in DNA, alter the cell cycle. Alterations lead the cell to proceed through the cell cycle with damaged DNA, which contributes to the kinds of chromosomal anomalies seen in Figure 2. These in turn lead to further deregulation of the cell cycle such that cellular proliferation is excessive, and apoptosis reduced. The net effect is an overabundance of cells, which is basically the situation in cancer.
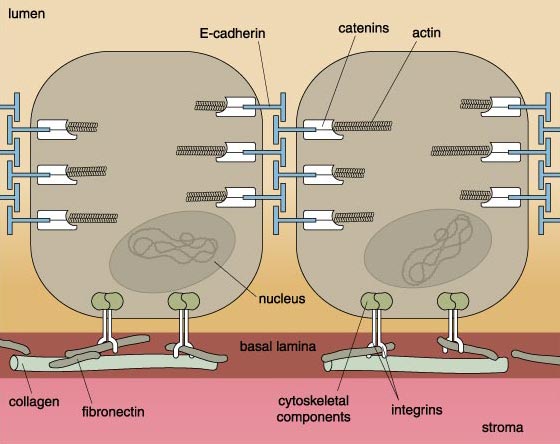
The impact of cellular adhesion in cancer is most clearly manifest in carcinomas—cancers of epithelial cells. Epithelial cells exhibit a rich variety of molecular interconnections. One of these is a very specific cell-membrane structure called the adherence junction. Primarily responsible for the junction is E-cadherin, a large protein that spans the membrane such that one end of the protein pokes out of the cell's surface, while the other end projects into the cell's interior. The external portion of E-cadherin forms interlocking bonds--like the teeth of a zipper—with E-cadherin molecules on neighboring cells. The internal portion hooks into the cell's interior protein skeleton, called the cytoskeleton. Recently, the proteins linking E-cadherin molecules to the cytoskeleton have been identified. These proteins are called catenins.
Aaron Cox
Before a cell can even start to move about, it first must break this complex, multidimensional interlocking structure. The adherence junction must be disassembled. This might be achieved in one of three ways. First, an alteration in the structure of E-cadherin could be introduced that would prohibit it from forming proper connections. Such an alteration would imply that the gene encoding E-cadherin had become mutated, corrupted in such a way that it encoded a less functional or even a nonfunctional protein. Surprisingly, mutations in the E-cadherin gene are relatively rare.
Second, there could be a decrease in the number of E-cadherin molecules on the cell surface. And, indeed, studies have shown that this is the case in many cancer cells. Finally, the linking proteins—the catenins—might be absent or nonfunctioning. This has in fact been found in an increasing number of cancers. The breakdown of cellular junctions not only diminishes the connections between epithelial cells, but it is also related to the cell's increasing internal disorganization.
It has long been noted that the internal architecture of cancer cells is irregular and disorganized. In general, E-cadherin expression is closely related to cellular differentiation. When they do occur, mutations in the E-cadherin gene often result in poorly differentiated cancer cells whose internal architecture no longer resembles the original structure. Furthermore, the lack of E-cadherin expression alters the relation of cells to each other. The same holds true for catenin mutations.
Of the three forms of catenin molecule, β-catenin has received the most attention. Work in the Johns Hopkins laboratory of Bert Vogelstein has shown that β-catenin not only helps to maintain tissue architecture, it also participates in the cell's internal chemical signaling system. Normally, β-catenin hooks up with another protein called APC (for adenomatous poliposis coli), whose function is to eliminate cells with mutations via apoptosis. A mutation in the APC protein leads to the growth of polyps, small benign tumors in the colon. For people who inherit mutations in APC, the risk is rather large that these polyps will become cancerous.
Aaron Cox
When acting alone, β-catenin has the potential to initiate cell division. The complex of β-catenin bound to APC prevents this from happening. In this way, cell growth is inhibited. Sometimes one or the other of the proteins in this complex becomes altered—owing to a mutation in the genes encoding them—and the complex cannot be formed. In that case, β-catenin, now unrestrained by APC, interacts with the cell's DNA and activates several genes that cause the cell to re-enter the cell cycle. Abnormal cell growth ensues.
Just as E-cadherins link cells to one another, molecules called integrins link cells to proteins such as collagen in the surrounding connective tissue. Like E-cadherin, the integrins span the cell's membrane, forming connections with connective-tissue proteins outside the cell as well as with proteins in the cell's cytoskeleton, inside the cell. One important difference between normal and cancer cells on the brink of metastasizing is that the cancer cells produce fewer integrins capable of linking with connective tissue than do the normal cells.
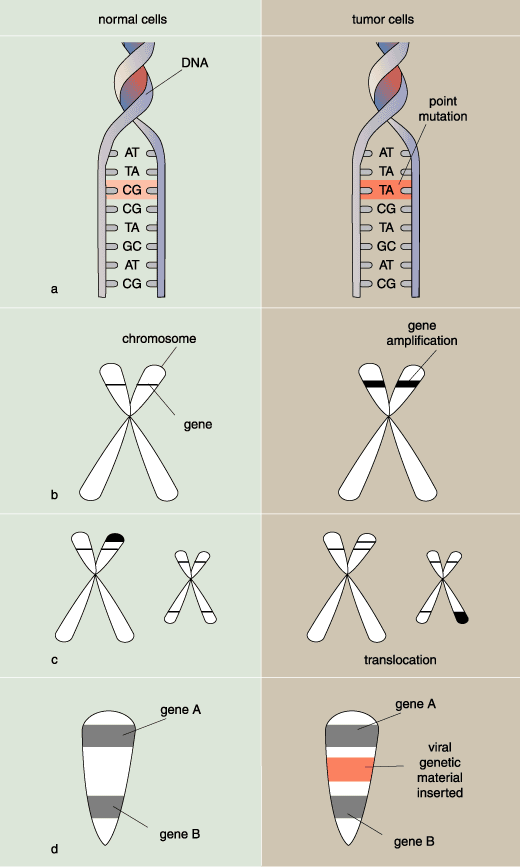
Interestingly, not all integrin synthesis stops. On the contrary: In cancer cells integrins are still being produced, but these constitute a set of integrin proteins different from those on normal cells. The so-called invasive cells on their way to becoming metastatic produce integrins that seem to help them migrate through the connective tissue or blood-vessel wall. Specifically, the migratory cells extend cytoplasmic protrusions (like the pseudopods of an amoeba) into the matrix of connective tissue. These protrusions latch on to proteins in the matrix with the help of the new integrin molecules and pull themselves through.
In order for a cell to make protrusions, its cytoskeleton has to be configured for motility. But as already noted, most cancer cells start out as sedentary cells and are neither programmed genetically for motility, nor is their basic skeletal structure conducive to it. Before a cancer cell can walk away from an organ, its cellular skeleton must be restructured for movement. And it is. The idea that a cell can alter its internal architecture is akin to suggesting that a fish can grow leg bones and walk off. And yet, biologists have long observed that cells do in fact change shape through the course of cancerous transformation.
In their normal state, epithelial cells come in several shapes—cylindrical, cuboid or flattened. Their cancerous counterparts become somewhat star-shaped and elongated and more closely resemble fibroblasts than they do epithelial cells. (Fibroblasts are the cells in the connective tissue that form a wall around epithelia and produce the matrix of connective-tissue proteins.) Interestingly, similar fibroblastic morphology is also observed in epithelial cells during embryonal development. This raises the interesting point that many characteristics of tumor cells resemble those normally seen only during the very early stages of embryonic development.
The internal cell structure is not the only thing that must be remodeled before the metastatic cell can leave its host organ. The matrix of proteins forming the connective tissue is like a wall without doors. Somehow, the metastatic cells must pass through this wall. This means that either the wall must be altered, or the cancer cells must acquire the ability to bore through tissue.
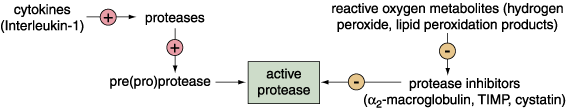
The latter scenario seems to take place. Cancer cells penetrate and pass through this barrier by dissolving it. For this purpose, cancer cells use proteases, enzymes that break down proteins, to degrade the mortar of the connective-tissue wall. It is probable that expression of active proteases is precisely coordinated in time as well as in extracellular location. For example, fibroblasts produce and degrade the matrix continuously, and under normal circumstances, this process is in equilibrium with other factors that rebuild the matrix. At the moment, it is not entirely clear whether the cancer cell releases its own proteases, or whether it stimulates protease release from other cells, or whether both processes take place sequentially or simultaneously. What is known is that certain messenger molecules alter the balance. It is possible that cancer cells have acquired the ability to synthesize these messenger molecules.
Aaron Cox
All proteases share a common feature: They are synthesized by cells in an inactive form, which must be chemically processed before they become active. Processing requires other proteases to chew away at a portion of the precursor protein, called a proprotein, in order to expose the active site of the protease. Complicating the balance is the fact that cells producing proteases may also produce natural protease inhibitors. Sometimes, chemicals in the environment can inhibit the protease inhibitors. So, the ability of a cancer cell to pass through its host tissue depends on some constellation of events that either promotes the synthesis and/or the activation of proteases or prohibits protease inhibition. The net effect is that proteases chew through the connective tissue, allowing the metastatic cancer cells to pass through. (Proteases secreted by endothelial cells may play a role in the angiogenesis required for the growth and metastasis of the primary tumor.)
Aaron Cox
There has been intense interest in learning more about the specific nature of these proteases, since inhibiting them would go a long way toward preventing metastasis. Much of the focus has been on the family of proteases called matrix metalloproteinases (MMPs). Members of this family can degrade various components of the connective-tissue matrix. The coordination and physiological functions of the different active forms of the MMPs are poorly understood. It is nevertheless clear that they are key to the mechanisms by which metastatic cells migrate through tissue compartments. In an in vitro model for malignant progression in human squamous cell carcinoma, one of us (Meade-Tollin), with coworkers at the University of Arizona at Tucson, has shown that three different MMPs have quite different expression patterns. We have observed that the expression of one of these, matrilysin, exists in higher concentrations in cells that form benign tumors, a presumed earlier stage in the progression to malignancy, than in cells that form invasive tumors in vivo. It is likely that different MMPs are involved at specific stages during which cancer cells become metastatic. Determining the levels of active MMPs in tissue could provide crucial information about the mechanisms of regulation of MMP activity.
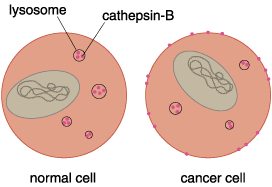
Two other protease families are also under investigation for activity during metastasis. The role of these families—cysteine proteases, such as cathepsin-B, and serine proteases, such as urokinase-type plasminogen activator—in cancer-cell invasion is less clear.
It is possible that members of some or all of these families of proteases interact during metastasis. One current scenario postulates that MMPs are secreted in their proprotein form by fibroblasts, but are activated by either cathepsin-B or urokinase-type plasminogen activator secreted by cancer cells.
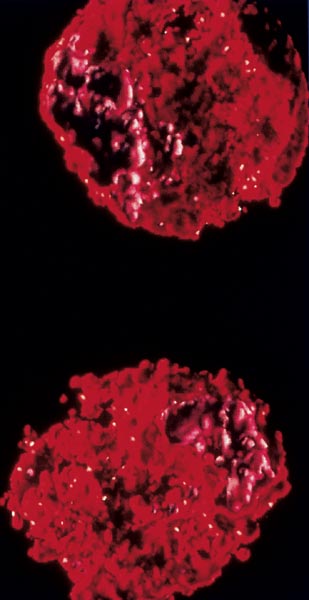
Digital image courtesy of Jan Van Marle, Academic Medical Center, University of Amsterdam.
The secretion of proteases, such as cathepsin-B, by cancer cells demonstrates another way these cells deviate from normal. Normally, cathepsin-B is sequestered within the cell inside membrane-bound vesicles called lysosomes. Lysosomes are the cellular trash cans that do double-duty as recycling centers. These vesicles contain enzymes—cathepsin-B among them—that can break down all macromolecules into their constituent units so they can be reused to make new macromolecules. Bonnie Sloane and her colleagues at Wayne State University School of Medicine in Detroit have discovered that cathepsin-B can be found in the extracellular environment of invading cancer cells. Furthermore, it appears that the invading cells not only secrete cathepsin-B, but can subsequently bind it to their surfaces.
Recently, one of us (Van Noorden) and colleagues were able to demonstrate that the surface-bound form of the enzyme is active. Once the metastasis is established at its new site, cathepsin-B continues to be expressed on the membrane, but in an inactive form. Urokinase-type plasminogen activator, like cathepsin-B, is present and active on invading cells. Such observations suggest that these proteases are at the head of a cascade of proteases that ultimately activate MMPs, which in turn chew up proteins of the connective matrix.
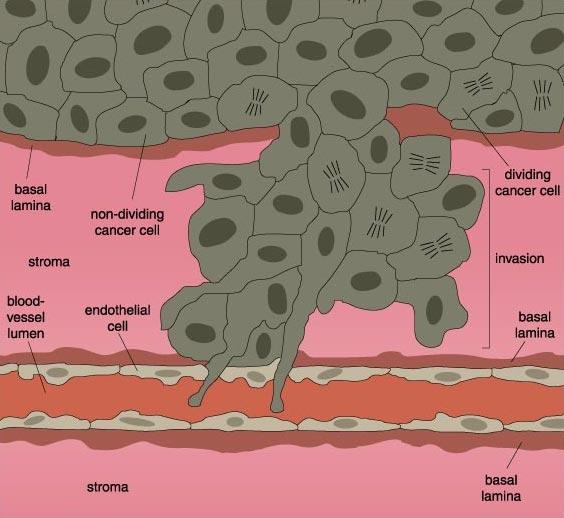
Metastasis is therefore a complex process, requiring the cancer cells first to release themselves of their intercellular adhesive bonds, then leave their cellular microenvironment and migrate through the connective tissue matrix encapsulating their tissue compartment in order to gain access to the blood vessels that carry them to a new organ. Once there, they must re-enact this process in reverse. They must pass through the blood-vessel wall into the new organ and there form new connections.
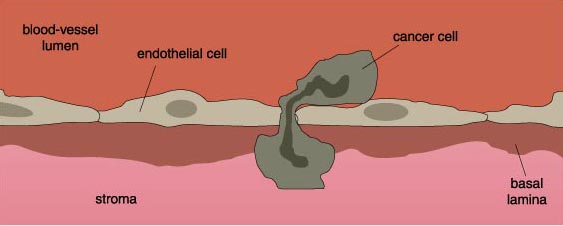
After it has successfully eaten through an organ's connective tissue, the cancer cell makes its way into a nearby blood vessel, squeezing between the endothelial cells lining the vessel lumen to enter the blood vessel itself.
Aaron Cox
Although the blood vessel provides a conduit for the metastatic cell to reach new tissues, the journey is fraught with peril—only 1 in 10,000 cells survives it. First, there is a large probability the cancer cell will be mechanically destroyed by the stresses that blood cells alone are designed to endure. In addition, the bloodstream is the very place where a cancer cell is likely to encounter the types of white blood cells—such as macrophages and natural killer cells—capable of destroying them, when the cancer cells are recognized as foreign.
Since cancers do take hold in new tissues, they obviously have developed mechanisms for transiting through the bloodstream. For example, they may travel in clusters, increasing the possibility that at least one of them will survive. Or they may surround themselves with blood cells such as platelets, which mask cancer cells from immune surveillance. (Platelets are blood cells involved in clotting.)
Cancer cells that survive the trip through the bloodstream ultimately home in on a new tissue. The selection of the new target is often quite specific to the type of cancer cell. For example, colon-cancer cells have a high affinity for the liver, whereas lung-cancer cells often metastasize to the brain, bones, adrenal glands and pancreas. The choice of target is quite likely determined by very specific interactions between molecules on the cancer-cell surface and molecules on the surfaces of the endothelial cells that line the blood vessels in the new host tissue.
Aaron Cox
The entire range of specific interactions is not yet known, but it seems likely that carbohydrates protruding from the cancer-cell surface become bound to a type of carbohydrate receptor on the endothelial cells called a selectin. Normally, the carbohydrate-selectin interactions are used by white blood cells that need to identify particular tissues to combat local infection. Apparently cancer cells can exploit this system as well. Each cancer-cell type expresses a different set of carbohydrates on its surface, which would be attracted to different selectin molecules. The specificity of these interactions helps account for the differential homing specificities of different types of cancer cells.
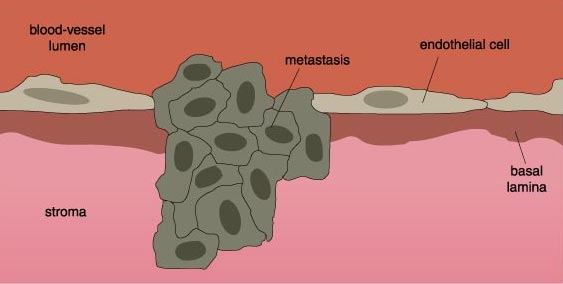
Once the cancer cell contacts a surface to which it can adhere, it rolls along the blood-vessel wall, propelled by the bloodstream, because the carbohydrate-selectin interactions are relatively weak. The cell comes to a complete stop as bonds, mediated by integrins, form between the cells. At this point, the cancer cell enacts a series of events that is almost the reverse of the events that allowed it to leave its primary organ. The cancer cell migrates into the host tissue by passing through the blood-vessel wall and degrading the connective-tissue matrix with proteases. The cancer cell is now ready to proliferate and form a new tumor in its new host tissue.
It is the fondest wish of all scientists who study cancer that it can be defeated. Scientists ardently hope to find some very precise way to curtail cancer's unrestrained growth without harming healthy cells, an advance that would represent a vast improvement over therapies currently available.
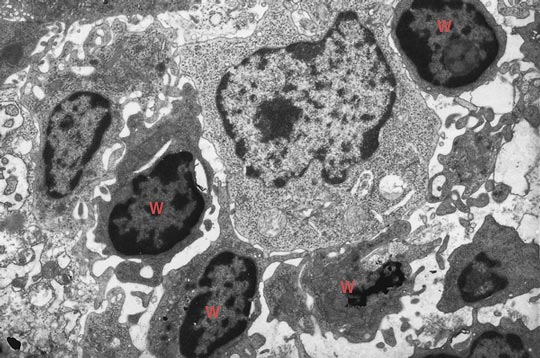
Photomicrograph courtesy of Patrizia Griffini, Academic Medical Center, University of Amsterdam.
Right now, when cancer is diagnosed, the tumor is often removed surgically. But there is always the possibility that some cancer cells remain at the original site, and that others may have already started to migrate to distant organs. So the patient is given radiation, which can eradicate cells by apoptosis. Radiation can be applied very specifically to sites in the body where the primary tumor was located in order to destroy any remaining cancer cells. But if undetected metastases are present elsewhere in the body, they go untreated.
For this reason, radiation is often given in conjunction with chemotherapy. The chemicals used in chemotherapy are almost all designed to curtail division and proliferation. The rationale is that cancer cells are dividing more rapidly than other cells in the body and are therefore most vulnerable to the effects of the chemotherapeutic agents. However, chemotherapy is quite a blunt weapon, and many normal cells with high turnover rates, such as skin, hair and blood cells, are affected along with the cancer cells. Sometimes cancer cells develop resistance to chemotherapy and become insensitive to its effects. Once that happens, the cell actually pumps out drug molecules, leaving itself and its progeny unharmed. Scientists therefore hope to target aspects of cancer cells that are different from healthy cells in order to develop drugs and therapies that attack the cancer cells only.
In the past decade several new therapies have sought to boost the patient's immune system. The approach grows out of the assumption that in cancer patients, the immune system is not able to effectively eliminate all of the cancer cells. The hope is that stimulating the immune system will increase the patient's ability to kill off cancer cells.
Aaron Cox
So far, this approach has been somewhat disappointing. In the test tube, experiments that combine immune and cancer cells are very definitely promising. The immune cells are effective in killing the cancer cells. But the situation is not repeated in vivo, where the complexity of whole-animal systems confounds the interactions between immune and cancer cells. Patrizia Griffini, in the Van Noorden laboratory, and other investigators have reported that in vivo, cancer cells do attract the attention of immune cells. Once the immune cells come face to face with the cancer cells, however, nothing seems to happen. The immune cells fail to launch the attack.
It seems that cancer cells have acquired the means to secrete large amounts of immunosuppressive messenger molecules, such as interleukin-10, transforming growth factor b and prostaglandin E2. Cancer cells also secrete molecules such as a2-macroglobulin, which scavenge immune-activating cytokines and cancer-cell-destroying proteases. In short, the cancer cells are extremely effective at manipulating their own microenvironments to their advantage.
Cancer cells may have an additional, and quite unexpected, effect on the immune cells that are supposed to kill them. Recent work by Jurg Tschopp and coworkers at the University of Lausanne in Switzerland demonstrated that the cancer cells may turn the tables on precisely those tumor-infiltrating immune cells deployed to fight the cancer. This work reveals that it is the cancer cells that in some cases are killing the immune cells.
Aaron Cox
Immune-killing cancer cells seem to have co-opted a mechanism usually employed by the killer immune cells. Almost all cells, including the cells of the immune system, carry a particular molecule on their surfaces called Fas. Fas is actually a receptor molecule that binds to another molecule called the Fas-ligand (FasL), which is normally expressed by immune cells. Under normal circumstances, immune cells hook their FasL molecule into the Fas receptor of the diseased cell. This interaction triggers a signal that causes the diseased cell to undergo apoptosis.
The Tschopp laboratory found that cancerous skin cells, specifically melanoma cells, also express FasL, whereas normal skin cells do not. Furthermore, these melanoma cells no longer express the Fas receptor. So when melanoma cells contact immune cells, they hook their FasL molecule into the immune cell's Fas receptor and cause the immune cell to undergo apoptosis. In the meantime, the melanoma cell is unaffected.
Current research is seeking to exploit such interactions to develop new anticancer therapies. For example, Claudia Friesen and her coworkers at the University of Heidelberg, Germany, found that the common anticancer drug doxorubicin enhances expression of both Fas and FasL on cancer cells. In effect, this drug causes cancer cells to kill themselves by inducing apoptosis.
Another promising class of anticancer drugs attempts to stop angiogenesis at the site of the tumor. Several recently characterized molecules, such as angiostatin and endostatin, have been shown to inhibit angiogenesis in laboratory animals. When inhibitors of angiogenesis are given to tumor-bearing animals, the tumors stop growing or, in some cases, even shrivel up. Several of these inhibitors of angiogenesis are in various phases of clinical trials and may prove to be potent against both primary cancers and secondary metastatic tumors alike. Such drugs, however, can potentially inhibit angiogenesis when it is wanted, for example, in wound healing. One solution to this problem may be to administer clotting agents selectively and directly to the tumor's blood supply, while leaving the normal blood supply unaffected.
Aaron Cox
Many recent experimental strategies against cancer attempt to compensate for or correct defective genes. The ras gene, for example, is mutated in a large number of human cancers. The Ras protein encoded by this gene is part of a pathway that responds to external growth signals by telling the cell to divide. The mutated gene encodes a protein that is permanently activated. That is, it is continually directing the cell to divide. One of the first steps in this pathway is catalyzed by an enzyme called farnesyl transferase, for which effective inhibitors have now been produced.
Other research strategies focus on restoring the function of growth-inhibiting genes. The favored strategy right now is gene therapy, which seeks to replace a mutated gene with a functioning one. Although this approach is likely to generate important therapies in the future, results so far have been disappointing. It is difficult to get the replacement gene to function properly and at levels that deliver therapeutic benefits.
In addition to efforts that seek to fight the primary tumor, much work has been focused on halting metastasis. Two main targets have provided the focus for new antimetastatic strategies. One target has been the adhesion molecules that link a cell to its original organ and that later allow it to attach to a new organ. The second focus for drug intervention has been the proteases that allow cells to chew themselves out of and into tissues.
As we have already seen, cells become unattached from their hosts by ceasing to make the adhesion molecules—such as E-cadherin—that connect them to surrounding cells. The obvious antimetastatic strategy would be to restore adhesion-molecule synthesis and prevent cancer cells from leaving their original site. One very active avenue of research involves gene-replacement therapy, in which functioning genes for the appropriate adhesion molecules could be delivered to cancer cells. This approach has worked in the test tube, but it is still far from being applicable at the bedside.
It may also be possible to modulate the relationship between integrins and the molecules to which they bind in the connective-tissue matrix. Integrins may be involved in angiogenesis as well as cell migration and the interactions between cancer and endothelial cells. For many integrins, the specific sequence of amino acids arginine-glycine-aspartate seems to be important for adhesion. Interventions have targeted this particular sequence, and have, in fact, been shown to reduce development of metastatic tumors in experimental situations. Still, these interventions are not yet ready for clinical applications.
Aaron Cox
Also interesting targets for drug intervention are the various proteases that allow metastatic cells to enter and exit tissues. Currently, only one inhibitor of MMPs, developed by British Biotech in Annapolis, Maryland, is in clinical trials. Typically, MMP inhibitors are not soluble in water, which limits their usefulness as drugs. But the new drug, called marimastat, is water-soluble. In animal models of cancer, marimastat strongly inhibits tumor development and metastasis and increases the animals' survival rate. Examination of the tissue of animals that have received this drug reveals an increase in the amount of connective tissue in and around the tumors as compared with untreated animals, suggesting that the drug is effective in halting the degradation of connective tissue by MMPs. This drug is even more potent in combination with the drug cisplatin.
The drug has been clinically tested on people in advanced stages of cancer. It was found to significantly increase survival with only the minor side effects of stiffness, local pain and discomfort. Protease inhibitors such as marimastat seem very promising for reducing the incidence of metastasis in patients once a primary tumor has been diagnosed.
Members of the Van Noorden laboratory are investigating inhibitors of other proteases involved in metastasis. We have recently been working with a specific small, water-soluble inhibitor of cathepsin-B, which seems to affect normal cells very little. We have found that the drug inhibits only the cathepsin-B on the cell membrane and not the internal lysosomal stores of the protease. This differential and complete inhibition of extracellular, but not intracellular, activity indicates therapeutic promise, because it blocks only the pathologically expressed cathepsin-B and not the physiologically required stores. We gave the drug orally to rats with colon cancer that had metastasized to the liver and found that the number of their tumors was reduced by one-third, and the size of the tumors was reduced by two-thirds. The study strongly supports the notion that cathepsin-B is involved in colon-cancer metastasis to the liver.
In the future, the question of how to treat cancer and its spread may take a back seat to strategies that prevent its development altogether. Scientists have increasingly discovered links between the environment, diet and the health of the individual. The hope in all of these studies is to eliminate the environmental dangers and educate individuals about how they can protect themselves and their bodies from agents that promote cancer.
Recently, much research has focused on dietary nutrients that might actually protect people from developing cancer. Among the agents under exploration are vitamins E and C, selenium, wine and substances from plants, called phytochemicals.
In the Van Noorden laboratory, we are looking at the relation between fatty acids, cancer and metastasis. We have found that omega-3 polyunsaturated fatty acids (PUFAs), which are found in fish such as salmon and mackerel, may help to prevent the development and progression of primary cancers, whereas omega-6 PUFAs from plants actually seem to promote tumor growth. It was recently demonstrated by us that omega-3 PUFAs inhibit proliferation of normal cells in vivo, whereas omega-6 PUFAs did not affect cell proliferation very much.
These effects can be explained by the formation of lipid peroxidation products that can damage DNA when it is uncovered during the cell cycle. Therefore, lipid peroxidation is kept to a minimum during the cell cycle of normal cells. However, lipid peroxidation products are generated in large amounts in cells that contain high levels of omega-6 and omega-3 PUFAs. Our investigations indicate that the inhibiting effect of omega-3 PUFAs on cell proliferation is mediated by lipid peroxidation products.
Since PUFAs seem to be important in preventing primary cancers, members of the Van Noorden laboratory were interested in seeing whether they had any effect on reducing metastatic tumors. Specifically, we explored the effect of PUFAs on colon cancers that metastasized to the liver in rats. Quite to our surprise, we found that, in rats, fish-oil treatment actually promoted the development of metastatic tumors in the liver. In fact, the results were dramatic. Fish oils caused a 10-fold increase in the number of tumors and a 30-fold increase in their size. Plant PUFAs did not affect the number of tumors, but their size was increased by 10-fold, as compared with animals on a low-fat diet. We believe the fish oils had the effect they did because the liver tumors were completely lacking the connective tissue known as stroma, which is normally found in and around tumors. It seems that tissues often try to encapsulate the tumor in stroma to limit its spread into the tissue. Our results would suggest that stroma formation is far more important in defending the tissue from cancerous invasion, at least in the liver, than we previously appreciated.
Our data also show that omega-3 PUFAs have at least two contradictory effects in relation to metastases in the liver. First, omega-3 PUFAs restrict cellular proliferation, as was shown in the first study. However, they also have the effect of inhibiting stroma formation in and around the tumors. This latter effect means that tumors can grow much faster in spite of the fact that omega-3 PUFAs inhibit cellular proliferation. We are now designing studies to investigate the effects of omega-3 PUFAs in the diets of colon-cancer patients at risk of developing liver metastases.
Until such a time that scientists become informed enough to prevent cancer, it is our hope that as processes crucial for the development of cancer and metastasis are ever more clearly delineated, this understanding will lead to more rational therapies. The new therapeutic interventions discussed hold great promise for slowing down or preventing cancer progression. The next generations of these drugs and strategies may even cure the disease someday. Since metastasis is the main cause of death in cancer patients, it will be a prime target for research to develop treatments that are now so desperately needed.
Click "American Scientist" to access home page

American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.