Zeroing in on Cancer
By Carlos F. Amabile-Cuevas
As targeted cancer therapies get more specific, the targets get rarer
As targeted cancer therapies get more specific, the targets get rarer

DOI: 10.1511/2010.86.366
Cancer can be viewed as a group of diseases linked by name but distinct from each other in ways that are immensely complex, or it can be considered as variations on a single theme. On the complexity side, we are now aware of myriad genetic changes linked to multiple hormones, receptors and signaling pathways that transform a normal cell into a malignant one. This transformation confers the ability to multiply without restriction, evade the immune system, extort nutrients from neighboring cells and migrate to other parts of the body.
Illustration by Tom Dunne.
From that perspective, tumors are no longer defined by the affected organ (lung cancer, for example), or even the type of cells in which they originate (say, nonsmall cells). Instead they are differentiated by the kinds of receptors they express—for example, positive or negative for epidermal growth factor receptors—or even by the mutations that keep a growth signaling pathway active when it shouldn’t be (such as the k-ras mutation). The variegated nature of cancer makes it impossible to develop a “unified model” for cancer biology, much less a unified therapy.
Despite these complications, a single feature distinguishes cancer cells and makes them fearsome: their unregulated reproduction. Most of the cells in the body of an adult achieve a differentiated state that defines them as neurons, muscle or blood cells, after which they no longer multiply. Cancer cells, on the other hand, actively duplicate themselves, often departing over generations from the strict program of differentiation to become exotic cells. The frenetic growth of cancer cells offered a target for the early versions of chemotherapy: Researchers could preferentially target cancer by attacking only cells in the process of cell division. Virtually all of the anticancer drugs that we call “cytotoxic chemotherapy,” from nitrogenated mustard to taxane drugs, act in this way. These drugs can significantly increase the lifespan of patients, but they come with terrible side effects, because some normal cells are in a more or less constant state of cell division. The epithelial cells that line the digestive tract must be constantly replaced. Hair grows without cease. Immune cells amplify in response to antigens. These cells are victims of the same assault designed to mow down cancer cells. The side effects of the chemotherapeutic agents are dramatic—sudden baldness, violent sloughing of the intestinal lining, immunosuppression—and they affect patients’ quality of life so deeply that chemotherapy is often more feared than cancer itself. Especially among patients of advanced age, there are some who would rather contend with cancer in their bodies for what time remains to them than endure chemotherapy.
Nearly 40 years ago, a new drug changed the way breast cancer was treated, opening a new front for attacking cancers in general. It had long been known that surgical removal of the ovaries interrupted the growth of breast tumors; hence it was proposed that estrogen produced in the ovaries promoted the growth of cancerous cells. The drug tamoxifen is an estrogen antagonist—it binds estrogen receptors without activating them while blocking the binding sites for estrogen. The great success of tamoxifen opened several new avenues in the treatment and diagnosis of cancer. First of all, it paved the way for research and development of other agents that interfere with estrogen signaling, such as aromatase inhibitors (aromatase is the enzyme responsible for estrogen synthesis), other estrogen antagonists, such as fulvestrant, and analogs of the gonadotropin-releasing hormone (which acts on the pituitary gland, starting a signaling cascade that normally includes estrogen synthesis and release). Tamoxifen and all other “hormonal” cancer therapies were the first “target therapies”—they were affecting features of the cancer cell much more specific than its unregulated growth.
Anti-estrogen drugs are only effective against tumors that have estrogen receptors, which amount to about a third of breast cancers. It was thus necessary to assess tumors for the presence of the receptor before starting anti-estrogen therapy. These new assays were the first attempts to adjust the drug therapy for cancer according to the cellular and molecular features of the tumor—beginning the era of “tailor-made” therapies.
Once the search began, other signals were found that induced the growth of tumors. One of the most important is epidermal growth factor, EGF. This molecule is critical during early development but is of little relevance in the adult body, so interfering with the EGF signaling cascade would be expected to have few side effects. However, it could arrest the multiplication of malignant cells that respond to EGF.
The EGF receptor is located in the cell membrane. We can imagine membrane receptors as counters at bureaucratic offices: an interface between the world at large and the inner world of red tape. An external signal such as EGF is like an application form received at the counter. After the application is received, a cascade of events starts; new forms are generated and passed through the office. In the cell, after receiving the external signal, the inner section of the receptor initiates a cascade by “stamping” other proteins, which activates them and sets the intracellular bureaucracy into motion. This biochemical stamping is often the addition of a phosphate group to a protein; the enzymes that add phosphate groups are called kinases, and the amino acid where the phosphate is most often added is a tyrosine, hence these bureaucratic gatekeepers are called tyrosine kinases. When the role of EGF in the promotion of tumor growth was demonstrated, researchers targeted the EGF signaling cascade for attack. Several drugs capable of inhibiting the tyrosine-kinase activity of the EGF receptor proved useful against different kinds of tumors. Although the side effects of the new drugs were much less severe than those of typical cytotoxic chemotherapy, these inhibitors also affected the tyrosine-kinase activities of other, unrelated receptors, adding a new variety of adverse reactions to cancer therapy.
Several advances in biotechnology led to improved assays and new kinds of therapeutic agents. On the diagnostic side, detection of receptors advanced from clumsy immunohistochemical tests to the exquisitely sensitive polymerase chain reaction and then to equally sensitive and massively parallel microarrays, which yield in a single assay a complete profile of receptors and mutations that can direct therapy much more precisely.
Our ability to engineer biomolecules opened the gate for designing antibodies and fusion proteins that can be used therapeutically, launching the era of “biological therapies.” The intentional production of antibodies is not new in medicine: Antitoxin sera and passive vaccines, in use for decades, are the result of injecting an animal with antigen, allowing it to create antibody in response, and then collecting the antibody from the animal’s blood and injecting it into a patient. This approach has an important limitation: The animal antibody is recognized as foreign by the immune system of the patient, which creates antibodies against the antibody. If administered a second time, the animal antibody will elicit a dramatic, even life-threatening response. But because they are molecules capable of recognizing other proteins in such a specific and mostly harmless way, antibodies were very tempting options for drug development. After injecting the selected antigen into animals and isolating the cells that manufacture the desired antibody, researchers replaced large chunks of the genes coding the animal antibody protein with human DNA sequences, keeping only the recognition sites responsible for the ”anti-antigen” function. This yielded humanized antibodies in which more than 90 percent of the antibody protein is identical to that in humans. These engineered antibodies are no longer antigenic in the host, and can be safely administered repeatedly.
Antibodies directed against EGF (for example, trastuzumab) or against EGF receptors (cetuximab) were developed to block the EGF signaling pathway, one or two steps before the tyrosine-kinase activity mentioned earlier. Targeting EGF reception at the cell surface avoids the consequences of inhibiting kinases inside the cell other than the ones activated by EGF receptors.
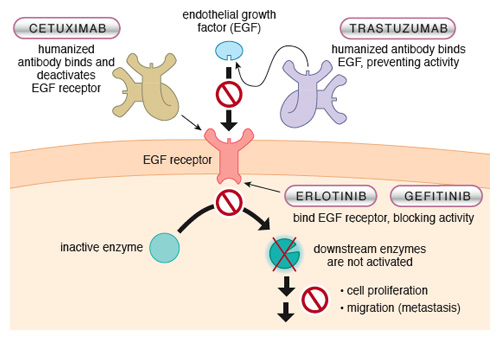
Illustration by Tom Dunne.
The clinical response to these anti-EGF therapies is very good, yet some tumors expressing EGF receptors do not respond to such agents. Why? The lesion in the signaling pathway may be further downstream. Imagine one clerk in the depths of a bureaucracy going mad and starting some process with no authorizing form stamped at the counter. One such mutation is k-ras, which is common in lung-cancer cells from Western male smokers. Drugs such as erlotinib or gefitinib that act as tyrosine-kinase inhibitors of EGF receptors do poorly in such patients, but are great drugs against lung cancer in Asian, female nonsmokers in whom the k-ras mutation is uncommon. Detection of k-ras mutations can predict the efficacy of anti-EGF therapies, an example of the high level of marksmanship made possible by target therapies.
Cancer is not just a group of cells growing in isolation; the physiological surroundings are also important. By affecting the microenvironment of cancer cells, we can sometimes restrain tumor growth. For example, a very small solid tumor can survive on oxygen and nutrients diverted from established blood vessels. However, as the tumor grows, some cells cannot get enough oxygen and start to suffocate—called hypoxia. In response, tumor cells synthesize a distress signal: vascular endothelium growth factor, or VEGF. This is another molecular “application form” that must reach another “counter” or receptor, but this receptor is not in the tumor. It is in the endothelial cells that form blood vessels. In response to the signal released by the tumor, endothelial cells start to grow new blood vessels (angiogenesis) to support the tumor with oxygen and nutrients. Without these new vessels, tumor growth is inhibited and the tumor is more susceptible to other chemotherapeutic agents.
As with EGF, VEGF has a marginal role in the body of an adult, where it is needed only to repair damaged blood vessels and fine-tune arterial pressure, so it is possible to interrupt this signaling pathway with minimal side effects. Anti-angiogenic agents now in use against a variety of tumors include bevacizumab, an antibody that binds VEGF and prevents it from reaching its receptor, and sunitinib and sorafenib, which are tyrosine-kinase inhibitors that interfere with kinase activity at VEGF receptors. It is believed that most solid tumors depend on angiogenesis to sustain their growth, so many molecules that affect this process are in the drug-development pipeline.
Another change in the tumor’s environment that might affect its growth is the inflammatory response cancers tend to elicit; our huge arsenal of anti-inflammatory drugs may thus play a role in the future of cancer therapy. Inflammation, particularly the chronic forms, has been targeted as a cause of cancer; tackling chronic inflammation or its underlying cause, often a bacterial or viral infection, might help to prevent cancer itself. Inflammation is the result of a complex signaling network that overlaps with other aspects of the immune response. It is therefore necessary to isolate the signals and pathways relevant to cancer and target them with specific drugs. Our current approaches to inflammations are still too coarse (inhibition of prostaglandin production or cytokine signaling), but new efforts using biological and even genetic therapies are currently being explored.
Despite the successes of target therapy, much remains hidden. Of capital importance are the mysteries of what allows cancer cells to migrate and colonize other organs—metastasis—and what we can do to stop it. Other possible targets include intercellular adherence, hypoxia response, tumor apoptosis (programmed cell death) and others. Most research is focused on the obvious growth signals (estrogen, EGF), but many tumors grow with no requirement for external promotion. We must search for targets within these cells as well. Because patient care takes priority over research, all of these new target and biological therapies are clinically tested as supplements to older chemotherapeutic schemes in trials that take several years; the usefulness of these new drugs on their own will have to be tested in the future. Unfortunately, there is a growing problem of patients showing less willingness to be enrolled in clinical trials. This reluctance can halt the progress from discovery to clinical use of even the most promising drug; it has been singled out as the most immediate challenge in the development of new cancer therapies.
Finally, there is the inescapable consideration of cost. Most new cancer treatments are very expensive. We may be tempted to remember how expensive the first biological therapy was: When used clinically for the first time, it was transported in armored trucks with police escort. The drug was even recovered from the urine of patients to whom it had been administered because that was less expensive than manufacturing it anew. This drug was penicillin 70 years ago, now cheaper than the glass vials it is sold in. But that is an oversimplification of the likely evolution of anticancer therapy, at least in the short and medium term. The truth is that market forces dramatically influence the way product research is now done.
As a society, we have put the discovery and development of new drugs in the hands of “Big Pharma.” As a result, profitability overrides health benefits. This is painfully obvious when we learn about highly effective drugs that never reach market because the illnesses they alleviate are rare. With tailor-made therapies, this will be an increasingly frequent outcome, as a particular target might be present in only a small fraction of cases. The brutal arithmetic of patient numbers, price and premarketing investment will determine whether a drug is pursued. This calculation may explain the absence of the spectacular cures that were predicted to follow the deciphering of the human genome.
Patients, then, are trapped between profit-driven research and the permanent drive to curtail healthcare expenses. In countries where healthcare services are financed and operated by the state—the prevailing model in many quite capitalistic European nations—a different balance decides whether a drug is approved for use. If the cost of the drug is more than the annual gross domestic product per capita multiplied by the number of years gained, on average, by the treatment, it is not purchased. This might sound like the brainchild of a sinister accountant at the World Bank, but it is also the way decisions are made about life-saving vaccines endorsed by the World Health Organization. In this way, drugs that may spectacularly benefit some patients are not used if the average patient does not respond so dramatically or if the country these patients live in has a low per capita gross product. This policy, in turn, discourages development of new drugs that may be rejected by this decision-making process.
We have the technology and the basic knowledge to embark on the search for individual genes and proteins that are responsible for essentially all types of cancers. This endeavor could enable target therapies that transform deadly tumors into manageable, chronic disease, if not offering outright cures. Such treatments would have to evolve together with precise diagnostic tools, so that tailor-made therapies could be assembled for each patient. But this fantastic perspective may end up crashing into the wall of economic concerns, which are the consequence of marketing health as if it were merchandise.
Click "American Scientist" to access home page

American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.