The Molecular Anatomy of an Ancient Adaptive Event
By Antony Dean
Protein engineering identifies the structural basis of a 3.5 billion-year-old adaptation
Protein engineering identifies the structural basis of a 3.5 billion-year-old adaptation

DOI: 10.1511/1998.17.26
Gradualism is a cornerstone of evolutionary theory. In the classic view, alterations to an organism or its parts accumulate slowly, imperceptibly changing form and function until gradually there emerges a being or an appendage or a cell or a protein that differs from the ancestral form. But gradualism is not the only game in town.
For a while now, there have been some upstart ideas, ones that suggest that most changes to the genes that ultimately direct the formation of beings do not matter much. Furthermore, alterations that do matter—changes that adapt an organism or one of its parts to perform a new function—happen in fits and starts.
In 1968 Motoo Kimura of the National Institute of Genetics in Japan proposed something radical for the evolution of proteins, the macromolecules that provide much of a cell's structure and do almost all of its catalytic work. Proteins evolve when alterations to the genes encoding them result in a change in the sequence of their constituent amino acids. Kimura suggested that most amino acid replacements that accrue during the evolution of proteins are the result of pure blind chance. Evolution without adaptation! Though radical, his idea was grounded in two well-understood phenomena.
The first phenomenon is the familiar process of random mutation. Each generation produces a new crop of alleles (genetic variants), most of which are not adaptive and are simply eliminated by natural selection. Kimura supposed that adaptive mutations are so infrequent that they can be ignored, and that the remainder are selectively neutral—they are neither favored nor disfavored by natural selection. These neutral alleles enter the gene pool.
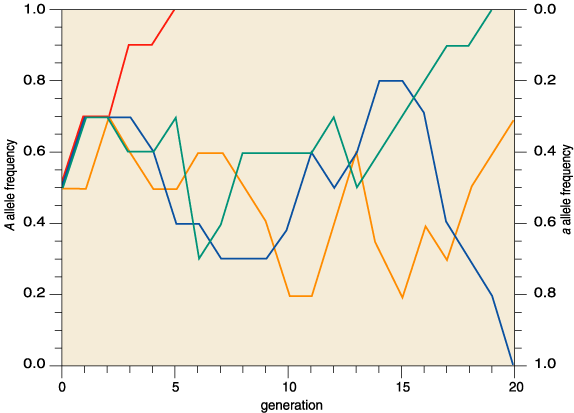
The second phenomenon, random genetic drift, is the tendency of gene pools to lose neutral alleles over many generations. Each generation is but a random sample of the previous generation's variants, and as such is subject to sampling error just as a Gallup poll would be. Over time, sampling errors accumulate: A lucky rare allele becomes common, or a common allele becomes rare, perhaps even lost from the gene pool (Figure 2).
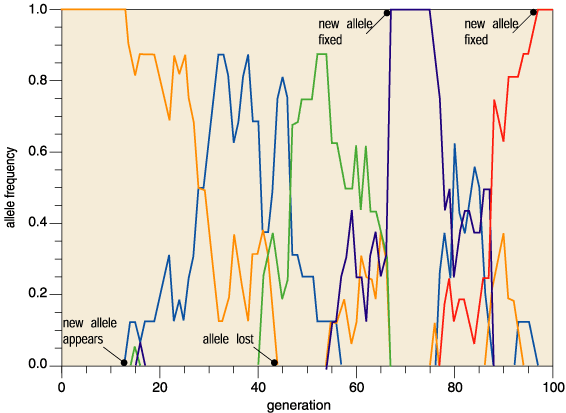
Kimura's radical idea pitted neutral mutation against random genetic drift. Mutation pumps new neutral alleles into the gene pool, while random genetic drift purges alleles from it. This churning flux of alleles causes neutral mutations to accumulate. Return to the gene pool in a million years and all the alleles have changed; in another million years they have all changed further still (Figure 3). Molecular evolution is a result of pure blind chance.
As a broad description of protein evolution this, the neutral theory of molecular evolution, has proved extraordinarily robust. After 30 years of intense research biologists have yet to provide a compelling picture of the molecular basis of a single ancient adaptive event. The difficulty arises from the very nature of the data collected. The linear sequences of amino acids in proteins, and of the nucleic acid bases in genes, are readily determined. By comparing the differences along these sequences it is possible to tease apart their historical relationships. Once determined, the neutral model can be subjected to various statistical tests. Unfortunately, the tests are so weak that rejection is possible only in extreme cases. Furthermore, minor modifications to the neutral theory frequently accommodate small deviations from naive expectation.
This is most frustrating, for even Kimura’s theory does not deny the importance of natural selection in evolution; its only claim is that adaptive mutations are rare, not nonexistent. Identifying rare adaptive amino acid replacements in an ocean of neutral change presents a formidable challenge.
Another place for biologists to start is with phenotypes. Phenotypes are the physical manifestations of genetic systems interacting with the environment. Although selection targets phenotypes (height, weight, eye color, catalytic efficiency, number of appendages etc.), only the genetic components are inherited by the next generation. It is perhaps for this reason that serious discussion of phenotypes rarely appears in studies of molecular evolution, which instead concentrate on the gene sequences. Yet phenotypes are crucial to understanding the process of natural selection because they show how organisms function in environments. If instead of merely analyzing gene sequences, evolutionists attempted to understand molecular phenotypes, the form and functions of proteins and enzymes in physiology and metabolism, they might gain deeper insights into the molecular basis of ancient adaptive events.
The challenge, then, is to return molecular phenotypes to molecular phylogenies, and in so doing to recover the molecular basis of truly ancient adaptive events. Here, I shall combine four approaches to do this. Each of the seemingly disparate fields of metabolism, phylogeny, x-ray crystallography and protein engineering makes its own very essential contribution to this story. None stands alone; indeed, their themes weave in and out, forming a tapestry rather than a simple linear string of arguments. An understanding of metabolism identifies the cause of selection, but the comparative methods of phylogenetics tease out the history. Establishing this history requires results from x-ray crystallography, results which also identify key amino acid replacements with important functional consequences. Techniques from protein engineering are then used to mutate these residues in an effort to show that they alone are responsible for the observed differences in phenotype.
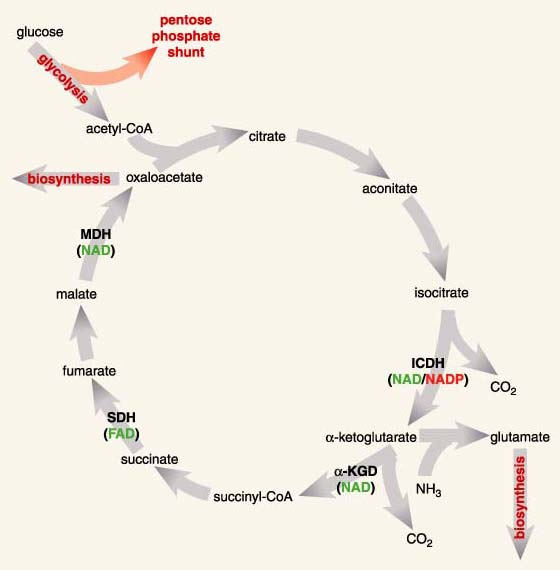
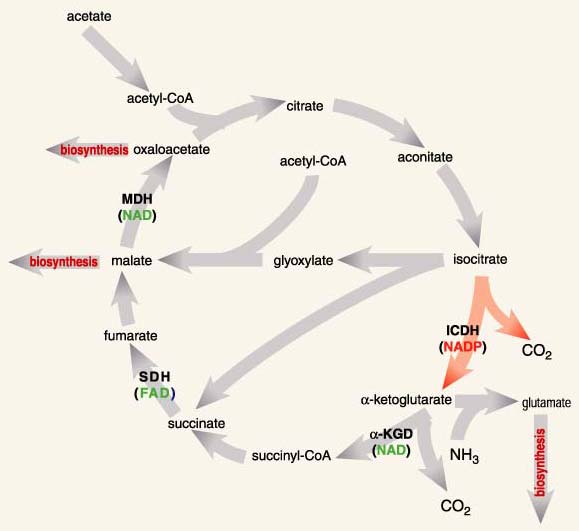
The phenotypes I am interested in belong to the isocitrate dehydrogenases (ICDHs), a family of enzymes found in an overwhelming majority of species. ICDHs catalyze a reaction central to energy production in the citric acid cycle and to the biosynthesis of glutamate, an amino acid. In fact, glutamate biosynthesis is doubly important because it is a primary means by which nitrogen, in the form of ammonia, becomes trapped in biomolecules. Organisms lacking ICDH must obtain energy through fermentation and glutamate from diet.
The specific chemical reaction catalyzed by ICDHs involves a reduction reaction—the transfer of a hydrogen from isocitrate to another molecule, called a coenzyme. The coenzyme will then cede this purloined hydrogen atom in the course of various other reactions necessary for life. In effect, the coenzyme is molecular money, part of a currency recognized by many enzymes, a currency that links and coordinates the various disparate parts of metabolism in much the same way that money greases the disparate parts of an economy.
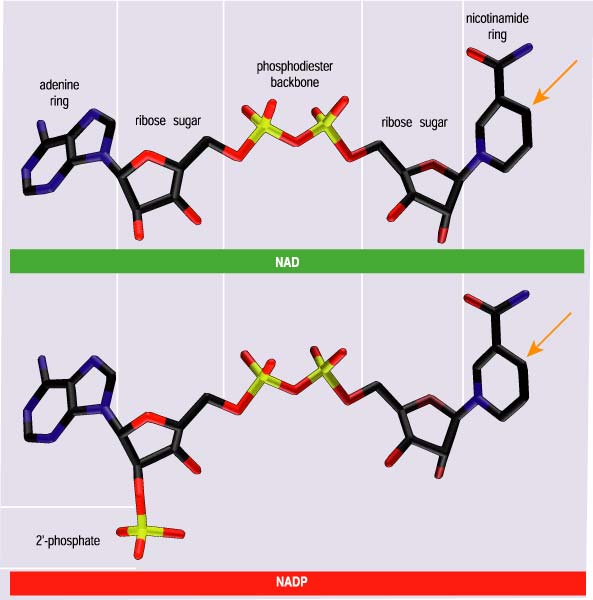
ICDHs reduce one of two coenzymes, nicotinamide adenine dinucleotide (NAD) or nicotinamide adenine dinucleotide phosphate (NADP). Despite structural similarities and identical catalytic chemistries (Figure 4), cells use NADH and NADPH (the additional H refers to the purloined hydrogen atom) in very different ways. NADH is used to synthesize adenosine triphosphate (ATP), another coin in the energy currency of life. NADPH is essential for numerous reactions that contribute to cellular maintenance and growth, including the synthesis of certain amino acids, such as glutamate.
All ICDHs display a marked preference for one of the two coenzymes, a preference that depends on metabolic role and cellular localization. NAD-dependent ICDHs are found in mitochondria, the powerhouses of eukaryotic cells, where ATP is synthesized. NADP-dependent ICDHs are found in the cytoplasm, where, among other things, they help reduce excess food to fat (pity). Even ICDHs from the single-celled bacteria, which lack the complex internal organization of eukaryotic cells—those found in plants and animals—display these marked preferences. Most members of the class of bacteria called eubacteria have an NADP-dependent ICDH, although a few have an NAD-dependent ICDH instead. The specific problem I shall tackle is the evolution of coenzyme usage in eubacterial ICDHs: When it happened, why it happened, where it happened and how it happened.
Mitochondria are the powerhouses of eukaryotic cells, where ATP is synthesized. Their ancestors were free-living eubacteria, until they were engulfed by a primitive eukaryotic cell some 3.5 billion years ago. Although most mitochondrial genes were lost long ago, a few, including those of the mitochondrial ICDH, were transferred onto the chromosomes in the eukaryotic nucleus. So, like modern eubacterial ICDHs, mitochondrial ICDHs are the direct descendants of an ancient eubacterial ICDH. However, unlike the majority of modern eubacterial ICDHs, which are NADP-dependent, all mitochondrial ICDHs are NAD-dependent. This immediately suggests that a switch in coenzyme usage took place very early in the history of life: Either the ancestral eubacterial ICDH used NADP, and NAD use by modern mitochondrial ICDHs is a derived trait, or the ancestral eubacterial ICDH used NAD, and NADP use by modern eubacterial ICDHs is derivative.
Any attempt to distinguish between these two competing possibilities lies firmly in the domain of phylogenetics, a field that seeks to reconstruct the genealogical relationships among organisms. The results of a phylogenetic analysis are usually presented in the form of a tree. The process begins by aligning the sequence of amino acids in a protein such that the amino acid positions in one ICDH correspond to those in another. Next, the molecular phylogenetic tree is constructed. The third step is to identify the root of the tree—the one position in the tree from which all sequences ultimately arose. And the fourth and final step is to interpret the tree in an evolutionary context.
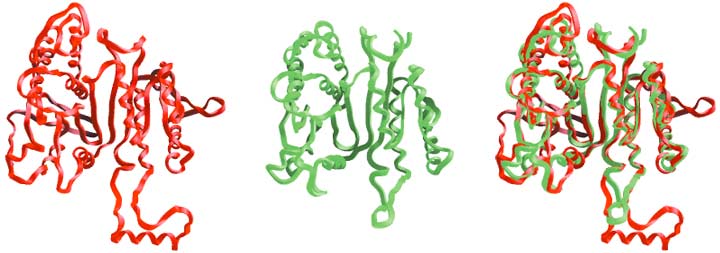
Most sequence alignments are based on some algorithmic criterion, such as maximizing the number of identical amino acids among the proteins. This works well for sequences that are similar, such as the eubacterial ICDHs or the ICDHs found in the eukaryotic cytoplasm (Figure 1). However, eubacterial ICDHs are so very dissimilar in their sequences from eukaryotic cytoplasmic ICDHs that little confidence can be placed in the alignments between these groups, no matter how powerful the computer program used.
Antony M. Dean
Fortunately there is another, exquisitely precise though little used, means to align sequences. Figure 1 shows a comparison of the basic frameworks of the ICDH from the bacterium Escherichia coli and the isopropylmalate dehydrogenase, or IPMDH, from the bacterium Thermus thermophilus. IPMDH carries out a similar reaction to ICDH in the biosynthesis of the amino acid leucine. Visual inspection reveals that the frameworks are quite similar. This is quite remarkable when one considers that only 20 percent of their amino acids are shared in common. On closer inspection many structural features, such as the flat sheets and the helices on the surfaces, obviously superimpose. There are some differences between these structures, though. For example, ICDH possesses a helix and a loop for which there is no equivalent in IPMDH. Here, a gap must be introduced into the amino acid sequence of IPMDH when aligning it against that of ICDH.
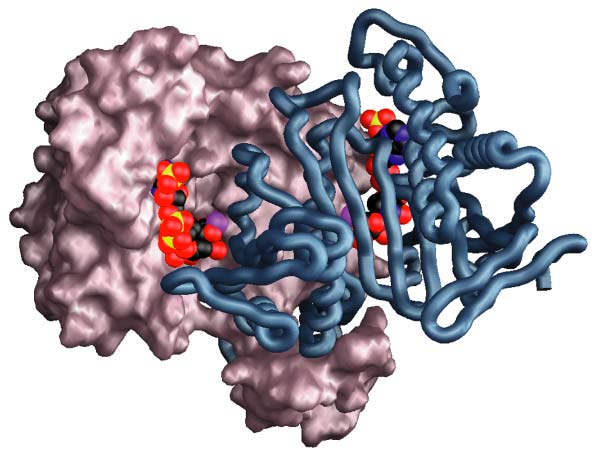
Enzymes catalyze reactions by binding and aligning the reagents—substrates and coenzymes—at specific sites on their surfaces called active sites (Figure 6). The specific amino acids (called amino acid residues when in a protein) crucial to the binding of both substrate and coenzyme and to catalysis in ICDH have been identified using x-ray crystallography (Figure 7). Being essential for function, these residues are necessarily highly conserved in the course of evolution. They provide key landmarks when aligning sequences from other distantly related groups, such as the mitochondrial and cytoplasmic ICDHs of eukaryotes, for which x-ray structures are not yet available. A knowledge of the three-dimensional structures of just two proteins, E. coli ICDH and T. thermophilus IPMDH, proved essential to the successful completion of the first step—obtaining a reliable alignment for this entire family of dehydrogenases.
The second step is to construct the phylogeny. There are a number of approaches to do this, each with its own adherents, even vehement supporters. Pheneticists construct phylogenetic trees based on the percentage of similarities between pairs of amino acid sequences. As an example, take the four amino acid sequences AKGCV, ALMSD, CLMQS and CGLCV, in which letters designate particular amino acid residues. These sequences can be grouped into two pairs, the first with the last and the second with the third, on the basis that sequences within a pair differ at three sites only, whereas sequences drawn from different pairs differ at a minimum of four sites. Thus, the first and last sequences resemble each other more closely than they resemble the second and third, which are deemed to form a distantly related group.
Cladists take a fundamentally different and perhaps a less intuitive approach, attempting to reconstruct the topology (branching order) of a tree using only shared differences. Of the five positions in these sequences only the first would be used by a cladist because the first two sequences "share" an A and the second two sequences "share" a C. In pairing the first sequence with the second and the third with the fourth, a cladist arrives at a rather different phylogeny than the pheneticist. Different methods sometimes yield different phylogenies.
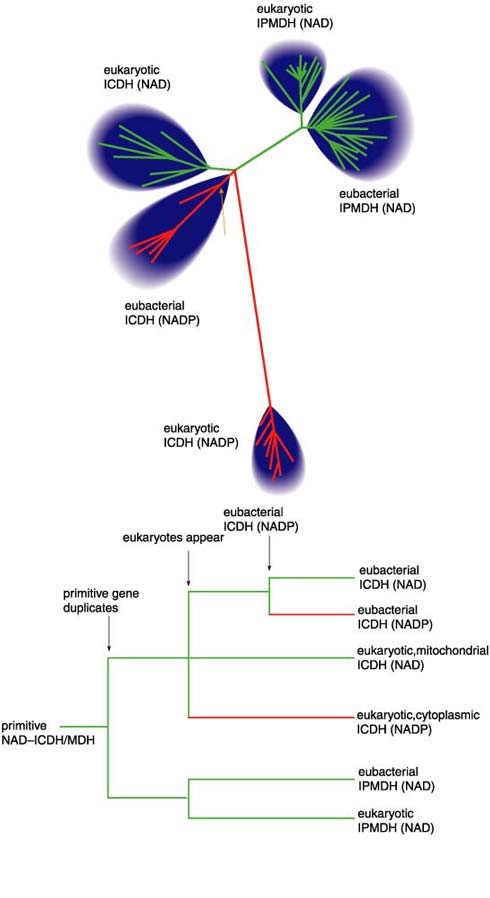
So which is the most reliable method for reconstructing phylogenies? The answer is unambiguous: We biologists do not know. So, rather than getting bogged down in the perennial debate as to which of the various methods is best, we use a variety of approaches (the phenetic techniques of neighbor joining and maximum likelihood and the cladistic technique of maximum parsimony) to recover phylogenetic trees that, within statistical error, are indistinguishable. All these trees consist of four distantly related groups of enzymes (Figure 8): the eubacterial NADP-dependent ICDHs, the eukaryotic mitochondrial NAD-dependent ICDHs, the eukaryotic cytoplasmic NADP-dependent ICDHs and the eubacterial and eukaryotic NAD-dependent IPMDHs. The four groups join at a central node. The precise pairwise branching order at this node is not resolved in these data.
As it stands, this tree is unrooted. There is no sequence that we can confidently assert should join the tree at its most ancient origin, the root. But we need a root because a root provides directionality in time: It tells us what is ancient and what is modern. Fortunately, a biochemical argument can be used to place a root on this tree. Ancient single-celled organisms must have synthesized all the amino acids essential for life because they could not have obtained them from diet—when life first began there was literally nothing to eat. They must have had an ICDH activity to synthesize glutamate, and they must have had an IPMDH activity to synthesize leucine. Therefore, the root of the tree lies on the limb joining the ICDHs to the IPMDHs. This completes step three.
The final step is to interpret the rooted tree (Figure 8) in an evolutionary context. An ancient single-celled organism had a single gene encoding a primitive enzyme that had the catalytic activities of both ICDH and IPMDH. The gene duplicated, and further evolution produced the first true ICDH and the first true IPMDH. Later, the eukaryotes emerged, an event reflected in both the ICDH and the IPMDH branches of the tree.
The tree also reveals that NADP usage in the ICDHs may have evolved independently twice, once in the eukaryote lineage and once in the eubacterial lineage. Both events took place around the time that bacteria were first taken up by primitive eukaryotes, which led to the modern mitochondria—some 3.5 billion years ago judged from fossil evidence. Thus, the order in which coenzyme usage evolved is resolved in favor of NAD dependence being ancestral to NADP dependence.
Antony M. Dean
But why did an ancient bacterial NAD-dependent ICDH evolve the ability to use NADP instead? As I have shown, such a switch represents a major shift in metabolic role, from energy production to biosynthesis. Adaptation to using acetate as the sole source of carbon and energy provides a possible explanation for the evolution of NADP dependence. All modern bacteria capable of growing on acetate generate the vast quantities of NADPH needed for biosynthesis in the citric acid cycle (Figure 9). E. coli, for example, uses ICDH to generate 90 percent of the NADPH necessary for growth on acetate. In contrast, bacteria retaining an NAD-dependent ICDH are incapable of growing on acetate. The switch by ICDH in coenzyme usage, from NAD to NADP, represents an adaptation, one of several in fact, to growth on acetate as the sole source of carbon and energy.
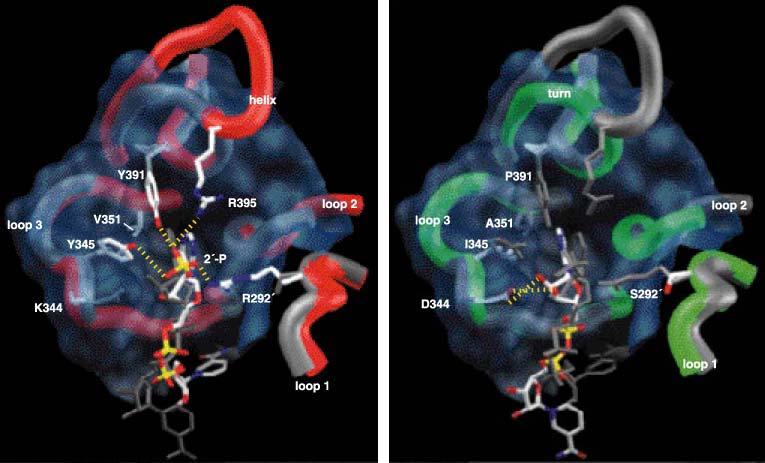
In order to catalyze reactions, enzymes must first bind the substrates and coenzymes in crevices on their surfaces, called active sites. The part of the active site that we are interested in is called the coenzyme-binding pocket, for it is here that ICDH and IPMDH bind and discriminate between NADP and NAD (Figure 10). The coenzyme-binding pockets of ICDH and IPMDH are constructed from three loops, with a helix in ICDH being substituted by a sharp turn in IPMDH. This overall similarity in construction is a reflection of common ancestry (the coenzyme-binding pockets of unrelated dehydrogenases are structurally distinct) and the need to bind coenzymes that are structurally very similar. The ability to discriminate between NADP and NAD depends on the properties of the amino acid residues lining the pockets.
Antony M. Dean
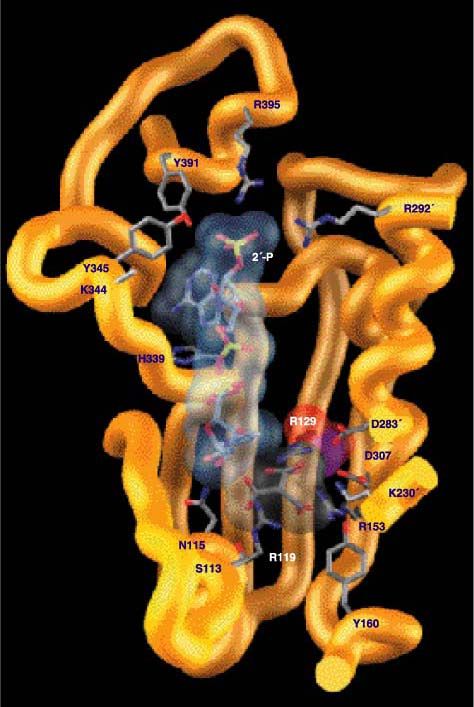
The processes of binding and discrimination are often likened to a lock and key, with an enzyme as a lock and the coenzymes (or substrates) as the keys. Only the correct key will fit snugly into the lock and allow a turn of the catalytic mechanism. The wrong key will not fit into the lock, or it will fit so poorly that turning the mechanism is difficult or even impossible. In this lock-and-key analogy, the amino acid residues lining the coenzyme-binding pocket are the lock’s tumblers. Just as real tumblers directly interact with the notches on keys, so these amino acid residues directly interact with chemical groups on the coenzymes. The coenzymes in question differ at only one group: In NADP an additional phosphate is attached to the ribose sugar at the 2' position, adjacent to the adenine ring (Figure 4). How amino acid residues lining the coenzyme binding pocket interact with the 2'-hydroxyl of NAD and the 2'-phosphate of NADP determines which of the coenzymes is used.
Three kinds of interactions are particularly important in making this determination: hydrogen bonds, electrostatic interactions and steric-packing effects. Hydrogen bonds are the same bonds that hold molecules of water together. They are sensitive to both the angle and the distance of the interacting atoms, which accounts for the ease of boiling water, whereby hydrogen bonds are broken to liberate free water molecules as gas. Electrostatic interactions occur between charged atoms. Oppositely charged atoms attract one another. For example, a crystal of common salt is held together by positively charged sodium ions interacting with negatively charged chloride ions. In contrast, atoms bearing similar charges repel one another: Static electricity makes hair stand on end as each charged strand repels its neighbors. Electrostatic interactions diminish with increasing distance, but remain insensitive to changes in angle. Steric-packing effects describe the physical fit of one molecule against another, much as the parts of a machine must fit snugly against one another.
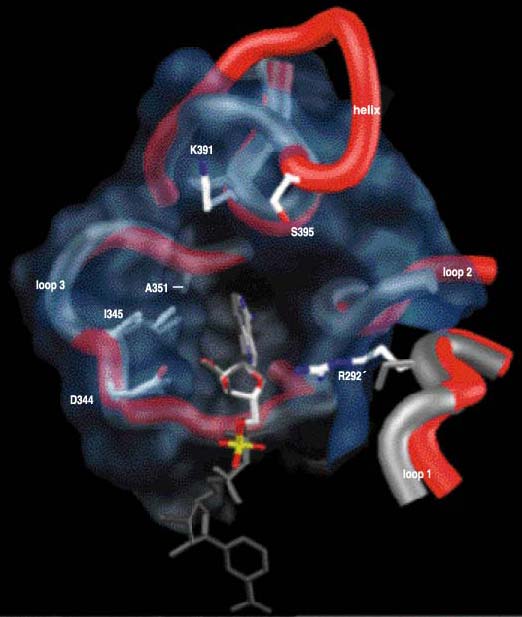
In E. coli ICDH, hydrogen bonds form between the 2'-phosphate of NADP and two residues of the amino acid tyrosine (found at positions 345 and 391), a lysine residue (position 334), and two arginine residues (positions 395 and 292', where the prime designates residues of the second protein subunit). The lysine and the two arginine residues are positively charged and so attract the negatively charged 2'-phosphate of NADP. It is the pattern of hydrogen bonds and the electrostatic attraction that stabilize the binding of NADP to ICDH.
These same five amino acids are replaced by other residues in the NAD-dependent dehydrogenases. Here, all favorable interactions with the 2'-phosphate of NADP are eliminated: The sharp turn of IPMDH has no site equivalent to 395 in the helix of ICDH, whereas serine (292'), isoleucine (345) and proline (391) are incapable of hydrogen bonding to the 2'-phosphate of NADP. New, smaller amino acid residues at sites 345 and 351 eliminate steric overlap with NAD, allowing it to tilt to the left, and the ribose pucker to flip. This brings the ribose hydroxyls sufficiently close to aspartate (344) that hydrogen bonds form, stabilizing the NAD bound in the pocket. Not only do these hydrogen bonds stabilize the binding of NAD to IPMDH, but the negatively charged aspartate actively repels the negatively charged 2'-phosphate of NADP.
The critical five amino acid residues that interact with the 2'-phosphate are conserved in all known sequences of bacterial NADP-dependent ICDHs. Furthermore, algorithms designed to infer ancestral sequences from a known phylogeny reveal that these same five amino acid residues were present at the earliest bacterial node (Figure 8). This suggests that adaptation to growth on acetate took place once in bacteria, and that the essential replacements have been preserved in the various diverse bacterial lineages for 3.5 billion years. Interestingly, none of these replacements is found in the cytoplasmic NADP-dependent ICDHs, where a large conserved insert, slap bang in the middle of loop 3 of the coenzyme-binding pocket (Figure 1), strongly suggests that an alternative means to bind NADP evolved independently in eukaryotes.
In contrast to the conservation seen in bacterial NADP-dependent ICDHs, only one of the five amino acids is conserved in all related NAD-dependent enzymes. This is the aspartate (344), so crucial to hydrogen-bond formation with the hydroxyls on the ribose-sugar portion of NAD. Alanine (351) and isoleucine (345) are occasionally replaced by other residues retaining two essential characteristics—that they be small and hydrophobic. A veritable riot of replacements can be found at sites 292' and 391, no doubt because these sites are no longer of any functional significance whatsoever to NAD binding.
That only six out of approximately 250 amino acid replacements at 320 positions (excluding all gaps) separating your typical bacterial NADP-dependent ICDH from your typical NAD-dependent enzyme are directly involved with determining coenzyme usage might seem reasonable to a protein chemist. However, it seems downright absurd to most evolutionary biologists brought up on a steady diet of pan-gradualism: many mutations, each of small effect, each improving the phenotype—just a little bit. Clearly, an experiment is in order.
The experiment is conceptually simple. The key amino acid residues of an NAD-dependent enzyme are introduced into E. coli NADP-dependent ICDH (which, recall, prefers to use NADP) using sequential rounds of site-directed mutagenesis, a technique of molecular biology whereby the amino acid sequence of a protein can be changed by introducing specific changes into the gene encoding it. The engineered gene then directs the synthesis of the engineered enzyme, which is then purified and its properties are studied.
Assuming the protein chemists are correct, the engineered proteins should change their coenzyme usage. Engineered ICDH, with the proper six amino acid replacements, should display a marked preference for NAD. Similarly, an engineered IPMDH should display a marked preference for NADP (wild-type IPMDH prefers to use NAD). A failure to switch coenzyme usage in either enzyme would indicate that additional, as yet unidentified, residues make significant contributions to coenzyme preference. Success in both cases would indicate that the key amino acid residues determining coenzyme preference have been correctly identified and that all remaining amino acid replacements separating NAD users from NADP users are of little or no significance with regard to this phenotype.
To determine the success f the experiment, we need a measure of the ability of an enzyme to discriminate between the two coenzymes. To do this we compare how efficiently each enzyme uses one coenzyme compared with the other. For example, it can be shown that wild-type E. coli ICDH uses 6,800 molecules of NADP for every molecule of NAD used, when both coenzymes are present in equal concentrations.
Five amino acid replacements in the coenzyme-binding pocket of E. coli ICDH shift preference away from NADP toward NAD by a factor of 106. However, the overall catalytic efficiency is far lower than that of a protein that evolved naturally to use NAD, the yeast mitochondrial NAD-dependent ICDH. This is not altogether surprising. The changes introduced to the coenzyme binding-pocket are quite drastic—whole chemical groups have been added, others have been eliminated, and the charge surrounding the pocket has been changed by four units. Optimizing activity may often require changes of a more subtle nature: an atom to be nudged a fraction over here, the angle of a hydrogen bond to be changed by 15 degrees over there, a minor steric clash to be eliminated. Such subtle changes can only be introduced by replacing amino acids at some distance from the coenzyme-binding pocket in the hope that their gross local effects diminish with distance. We tested this hypothesis by introducing bulky amino acids, to force conformational changes, at six sites surrounding the active site of ICDH. Two mutants combine to increase catalytic efficiency by a factor of 16, bringing it in line with that of the yeast enzyme (Figure 11).
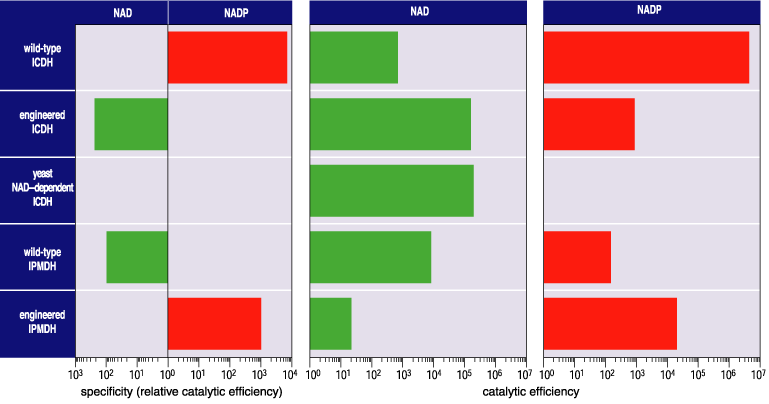
Both the engineered E. coli ICDH and yeast mitochondrial ICDH have lower overall catalytic efficiencies than wild-type E. coli ICDH. This is because five hydrogen bonds and three charge interactions allow NADP to bind more tightly than NAD, which instead is stabilized by a double hydrogen bond. On the other hand, the engineered ICDH has a higher specificity for its new coenzyme than does the naturally evolved IPMDH. Moreover, x-ray analysis reveals that NAD occupies precisely the same position in engineered ICDH that it does in wild-type IPMDH. By any measure, a highly specific NAD-dependent ICDH has been engineered. The last time such an enzyme used NAD in preference to NADP was 3.5 billion years ago!
Well, if you can engineer one way, you should be able to engineer the other way as well. However, engineering the coenzyme specificity of T. thermophilus IPMDH is far trickier than engineering ICDH. Now we must engineer the very architecture of a protein. The sharp turn of IPMDH must be replaced by a helix and loop so as to introduce an additional arginine residue, (395), to form a hydrogen bond with the 2'-phosphate of NADP (Figure 10). A 13-residue sequence modeled on the helix and loop of E. coli ICDH, but containing additional amino acid replacements to ensure that the insert lies correctly against the remaining protein, replaced the seven residues comprising the sharp turn in IPMDH.
Antony M. Dean
Together with four direct replacements lining the pocket (sites 292', 344, 345 and 351), a shift in preference from NAD to NADP by a factor of 100,000 was generated. The resulting mutant IPMDH contains the entire suite of amino acids found in ICDH, has a specificity for NADP of 1,000 and is twice as active as the wild-type enzyme (Figure 11). A highly specific NADP-dependent IPMDH has been engineered. The last time an IPMDH used NADP in preference to NAD was, well, never. All available evidence is consistent with the hypothesis that this engineered phenotype is unique in the entire history of life.
If it is difficult for many evolutionary biologists to contend with the idea that only six of approximately 250 amino acid replacements determine coenzyme usage, then their shock borders on disbelief when they learn that Steve Holbrook and his colleagues at University of Bristol changed a lactate dehydrogenase into a malate dehydrogenase by replacing just one amino acid, chosen from approximately 230 differences. This provides additional evidence that the traditional notion of gradual phenotypic change is quite clearly wrong. Major shifts in enzyme function may often require no more than a few changes.
The discovery that functional differences between members of a protein family are determined by only a few amino acid replacements is liberating. Freed from the assumption that such differences are determined by many replacements, each of small effect and all engaged in an orgy of horrendously nonlinear interactions, we come to the realization that the problem is far simpler than most of us ever imagined. We can study the history of natural selection. We can reconstruct the molecular basis of adaptations that first appeared in organisms long since extinct.
The realization that so few amino acid replacements determine function also helps explain a striking pattern of molecular evolution. Phylogenetic analyses reveal that major shifts in enzyme function are relatively infrequent, so that the evolution of enzyme function is characterized by (and with apologies to all paleontologists) a sort of punctuated equilibrium—long periods of stasis interspersed by brief periods of rapid phenotypic change. The shifts are infrequent because the interim periods are presumably characterized by strong stabilizing selection.
For example, cellular demand for NADH remains an unlikely explanation for the NAD dependence of IPMDH, particularly since its gene expression is regulated by the availability of leucine. Rather, NAD dependence has been retained because selection eliminates mutants of intermediate specificity—these, as protein engineering has shown, have lower overall catalytic efficiencies. The adaptive shifts are rapid because so few amino acid replacements are necessary to change function. Hence, an understanding of metabolism, and the relations between form and function in enzymes, combine to provide a ready explanation for a commonly observed pattern in molecular evolution.
Having found an adaptive explanation for half a dozen amino acid replacements, we might reasonably ask what the remaining 244 are doing. Some are undoubtedly involved with other major phenotypic adaptations, such as specificity toward isocitrate and isopropylmalate, thermostability, quaternary structure and regulation. For instance, the eukaryotic mitochondrial ICDHs consist of four subunits, two of which contain active sites and two of which carry out regulatory functions. Being overly generous we might ascribe 20 amino acid replacements for each major phenotypic change and still be left with a vast number of replacements of no apparent phenotypic consequence. What of these?
Many may be of no functional consequence, and hence selectively neutral. Many others may be involved with modulating enzyme function and hence be of adaptive value from time to time or from place to place. For example, one of the amino acid replacements remote from the active site in engineered NAD-dependent ICDH improved activity twofold and might reasonably be considered to fall into this category. Such treadmill replacements, selected at one time or another as evolving populations track environmental changes, do not alter the overall function of enzymes in any significant way.
Biologists will never be able to ascribe to each and every mutation accrued during the vast history of evolution a cause, be it adaptive or neutral. But as these studies show we can understand why, when and how certain mutations were adaptive, and thereby enrich our understanding of the molecular basis of evolution.
Click "American Scientist" to access home page

American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.