One Family's Search to Explain a Fatal Neurological Disorder
By Nissa Mollema, Harry Orr
With their help, researchers are advancing the 70-year effort to understand—and treat—hereditary ataxia.
With their help, researchers are advancing the 70-year effort to understand—and treat—hereditary ataxia.

DOI: 10.1511/2013.105.442
Ten-year-old Henry Schut noticed that his father was having increasing difficulties with his daily chores on their farm in rural Minnesota in the early 1900s. His father grew highly agitated by his display of clumsiness as he attempted to hook up the horses for plowing. Henry soon learned why his father was so distressed.
People were whispering about a mysterious disorder that plagued his family: About half of the children born into it developed symptoms of the disease in adulthood. Affected individuals first experienced walking difficulties, then hand incoordination, then slurred speech, and finally, labored breathing. The disorder was lethal within around 10 years of the onset of symptoms.

Photographs courtesy of the Schut family.
True to this distressing history, Henry's father's condition quickly progressed until he was extremely debilitated. Henry, along with his three younger brothers and one sister, was running all of the farm operations before their father passed away in 1924 at the age of 46. Several of Henry's uncles also passed away within the next few years because of the disease. Henry and many of his cousins were suddenly growing up in single-parent households. As life moved on after the family tragedies, Henry could not stop thinking about his father's condition. His eight aunts and uncles had many children, and he knew his siblings and cousins could eventually become ill with the disorder. He wondered what it was and who might be affected by it. He wondered if there was a cure; he wanted to prevent such a tragedy from happening again within the family. If he married and began a family of his own, he was concerned that he could pass the illness to his children, but there was no way for him to know.
Years after Henry's father and uncles had passed away, Henry's fears began to be realized: One of his close cousins, Bert, was showing early signs of the disease at age 20. Consultations with their local medical doctors proved extremely unhelpful in deciphering the disorder. In Henry's small town, rumors were spreading about the possibility that the family illness was contagious. At Henry's encouragement, Bert made the trip from western Minnesota to the university hospital in Minneapolis. The family hoped that medical specialists could give them an accurate diagnosis and possibly provide a treatment.
During this trip in about 1930, the doctors diagnosed Bert with ataxia. This diagnosis unified under one umbrella those affected by similar symptoms, but they still did not know what caused the disease, how to identify those who would be diagnosed later in life, or how to cure it.
Once diagnosed, family members researched what ataxia was. Ataxia is derived from the Greek term for "lack of order," and means having little control over muscle movements. Patients with ataxia walk with a wide stance and shuffling, small steps in an attempt to maintain balance. Their fine motor skills are also lost: They struggle to bring an outstretched finger to touch their noses. Henry's father lost the ability to tie his shoes, dress himself, and feed himself as the disease progressed. Ataxia robbed him of his ability to control both his fine and gross muscle movements, making each motion challenging and inefficient.
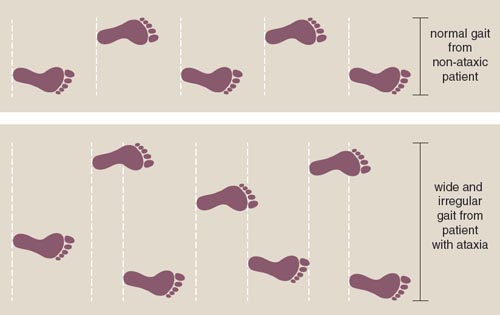
Illustration by Barbara Aulicino.
A variety of causes can result in ataxic symptoms. People who have had too much alcohol display temporary ataxia—they stagger and have difficulty keeping their balance. Permanent forms of ataxia, in contrast, can be debilitating and sometimes deadly. A traumatic brain injury, such as stroke or head trauma, may trigger ataxia. As the patient recovers from the injury, the ataxia may improve. More complex forms of permanent ataxia are caused by genetic mutations. In these cases, mutations that cause the disorder are passed from parent to child. Sporadic cases of ataxia, in which a patient with no family history of the disorder is diagnosed with the disease, are more mysterious. Hereditary and sporadic ataxias currently affect roughly 150,000 people in the United States and are typically progressive in nature, making the individual more disabled as the disease advances.
Whatever factor caused the disease became more toxic to the body as it passed from parent to child.
The doctors at the University of Minnesota took a particular interest in the ataxia that ran in Henry's family during Bert's visit to the hospital. At the time, the only form of hereditary ataxia that was known was Friedreich's ataxia, which was described in 1863. The pattern of inheritance seen in Henry's family was different from that seen in Friedreich's ataxia. Unlike Friedreich's ataxia, the ataxia in Henry's family affected members in each generation, instead of skipping generations.
Family members with ataxia also presented with a variety of symptoms that differed from Friedreich's ataxia, suggesting that they had a form of ataxia that had never been diagnosed. In this new ataxic disorder, slow, progressive symptom onset began between the ages of 20 and 30. Patients usually noticed problems with their lower limbs first and began having difficulty keeping their balance while walking. The university doctors suspected that the symptoms of ataxia were the result of degeneration in the brain or spinal cord.
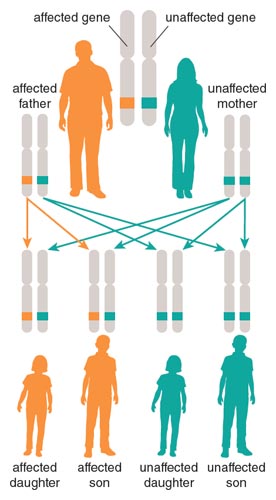
Illustration by Barbara Aulicino.
To better understand the genetic nature of the disease, the doctors asked Henry to create a family tree documenting which individuals were affected or unaffected by the disorder. The Schuts' family tree supported the fact that only family members with ataxia could pass the disorder to their children and that the disease displayed a dominant mode of inheritance (50 percent of children were affected).
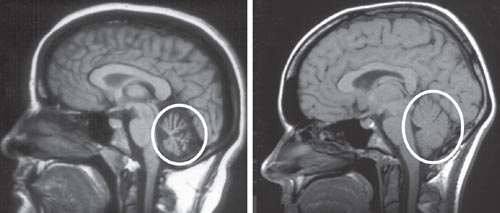
Although the family now had a diagnosis for the disease and a better understanding of how it was inherited, no one had information about the cause of the disease or how to treat it. Clinicians advised the affected family members to eat well and exercise, yet this did little to prevent progression of the disorder. For 10 years, Bert continued to become more disabled, until he passed away from ataxia in 1939 at the age of 30. At the encouragement of physicians, Bert's brain was sent to University of Minnesota to be studied. As doctors had previously predicted, extensive degeneration was seen within the spinal cord, which controls involuntary functions, and within the cerebellum, which controls voluntary movement. Physicians named the disease spinocerebellar ataxia type 1 (SCA1) because of the regions involved. But they still had no treatment or explanation for why it was occurring.
Henry dreamed of attending medical school to study ataxia and help his family. However, financial hardships during the Great Depression forced him to abandon his efforts to obtain a college degree. He returned home to the farm in 1934 and married in 1935. His cousin Ed also returned home to be closer to family after he was diagnosed with ataxia. He was a willing research participant and worked with Dr. Rossen at St. Peter's Hospital, where he donated cerebrospinal fluid for research. He was even directed to consume calf brains in hopes of identifying a potential treatment. Unfortunately, these efforts did not improve his condition.
Images courtesy of University of Washington Neurogenetics Clinic.
Henry's younger brother, John, made it his goal to find a cure for the family disorder and studied to become a medical doctor. He began practicing as a neurologist in the mid-1940s. Dr. John, as family members called him, completed a comprehensive study of the ataxia that ran in his family, which was published in Archives of Neurology and Psychiatry in 1950. This article described the symptoms of the disease, the multigenerational nature of the disorder, and an interesting phenomenon regarding the ataxia in his family—family members were showing symptoms of the disorder at earlier and earlier ages, an effect called genetic anticipation. Genetic anticipation is common in some hereditary neurodegenerative disorders, such as Huntington's disease, but at the time no one knew why this accelerated timing would occur. In the hereditary ataxia that affected the Schut family, each generation that inherited ataxia was more severely affected by the disorder. Whatever factor caused the disease became more toxic to the body as it passed from parent to child.
In 1949 Dr. John returned to a medical residency program to become a certified neurologist. Meanwhile, his older sister, Elsie, had become severely affected by ataxia, and he was determined to do all he could to help her. With each minor illness, her lungs became filled with fluid that her body was unable to expel. She coughed, gasping for breath, and Dr. John frantically aspirated her lungs so that she could take another gulp of air. Her death in 1953, at age 36, was especially difficult for him; he had done everything he could to keep her alive. Now Dr. John was even more determined to identify the cause of ataxia, and time was beginning to run out: He himself was showing symptoms of the disease. He suspected that there must be a genetic factor. This idea was based on the observations that multiple generations within the family were affected and that the disease symptoms increased in severity as ataxia passed from generation to generation. If he identified the factor underlying the hereditary ataxia by finding genetic association, he could potentially treat the disorder.

Photographs courtesy of a family that wishes to remain anonymous.
Such research called for resources, so in 1957 Henry and Dr. John founded the National Ataxia Foundation to raise funds. Ataxia was nearly unheard of, so raising awareness about the disorder was necessary. Dr. John worked with another relative to educate the public about ataxia. Soon, he secured private and government funding to conduct genome association studies. At the time, no other families with SCA1 were known, so the studies focused only on affected members of his own kin.
Dr. John examined blood and cerebrospinal fluid from affected and unaffected individuals, but he did not find a blood factor associated with the disorder. However, in the cerebrospinal fluid from ataxic patients, he saw higher levels of compounds attributed to death of neuronal cells. These compounds were possible by-products of the disease, but alone they could not explain the cause of ataxia. Dr. John kept studying ataxia but made little progress. At the time, science lacked the necessary tools to identify the factor causing the disorder.
As more cousins became disabled by ataxia and his own illness worsened, the lack of answers was increasingly frustrating for Dr. John; conversely, the possibility of identifying genetic underpinnings became ever more tantalizing. Without a genetic test, it was impossible to determine whether a family member was affected by the disorder. The typical age of SCA1 onset corresponded perfectly to the period in an adult's life when he or she is considering marriage and children. If a family member reached 33 years of age and remained symptom-free, then it was unlikely he or she would develop ataxia. Otherwise, he or she risked passing the disorder to the next generation. On the advice of Dr. John, at-risk family members who had a parent with ataxia delayed having children or avoided conceiving altogether.
Dr. John's ataxia worsened to the point where he was unable to conduct his research. This infuriated him and made him determined to do everything he could to stay alive to focus on finding a cure. His spirits brightened when Henry's son Larry entered the University of Minnesota Medical College in 1958. Henry had remained unaffected by the disorder, so his children were at no risk of developing ataxia. Yet he and his children were closely tied to the disorder, because three of Henry's four siblings were affected. Antibiotics and new medical interventions kept Dr. John alive for longer than any other family member with the disorder; he eventually passed away at age 51 after fighting ataxia for 23 years.
The field of ataxia research remained quiet for about 10 years. Dr. Larry Schut was practicing medicine in Golden Valley, Minnesota, but in the 1970s he received a phone call from the National Genetics Foundation regarding two brothers in Sioux Falls, South Dakota, who had a rare and incurable disorder. This phone call resulted in the diagnosis of 12 more people afflicted with SCA1 and the opening of several clinics sponsored by the National Ataxia Foundation. With a larger group of people to contribute to the research, Larry felt that there was new hope of identifying the underlying cause of SCA1. The Schut family assembled during a reunion, and Larry collected blood from the affected and unaffected members in attendance. In continuation of the genome association studies started by his uncle, Larry collaborated with Dr. Elving Anderson at the University of Minnesota. In 1984 they found a genetic region correlated with SCA1 located on chromosome 6. The region was near the human leukocyte antigen (HLA) complex, an area of the genome filled with many genes related to immune system function.

Image courtesy of the Orr lab.
That same year, Larry recruited the help of one of us (Harry Orr) to zero in on the genetic region with which SCA1 was associated. At the time, Orr had been studying the HLA complex since 1976, was also trained in neuroscience and genetics, and had an interest in the SCA1 research. The Orr lab was well equipped to analyze the genetic region associated with SCA1, and they began work on genetic mapping of the region in 1985. Genetic mapping during these early days of molecular research was challenging, labor intensive, and limited by an incomplete understanding of the human genome. Larry helped by providing blood samples from family members and giving us a detailed family tree.
Our lab then used restriction fragment length polymorphisms (RFLPs) to reduce the genetic region in question. In RFLP mapping, size differences in DNA of the same region are identified by cutting at specific sequences with the use of particular enzymes. If a size difference was noted between affected and unaffected individuals, that region of the DNA was further scrutinized to see how the sequences differed. After almost 10 years of work on the project, we were able to reduce the genetic region in question down to a more manageable span that was associated with the disorder.
At this point we were close to a breakthrough, but we needed help to quickly hunt down the specific sequence of DNA responsible for causing ataxia. A phone call from Huda Zoghbi of Baylor College of Medicine, who was investigating the genetic cause of ataxia in a different family, started a 20-year scientific collaboration with us that still flourishes. Several Schut family members also volunteered their time by working in the lab, and Larry continued to collect samples and update the family tree. In 1993, our collaborative efforts paid off: A stretch of DNA that was much longer than the same stretch in unaffected individuals was isolated by RFLP mapping.
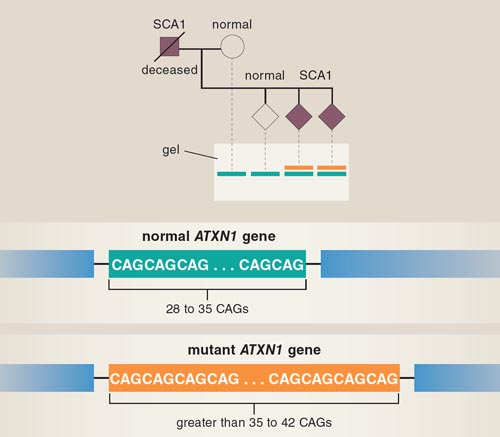
Illustration by Barbara Aulicino.
Subsequent analysis showed that the three-base sequence CAG, which codes for the amino acid glutamine, was repeated at this location in those with ataxia. Normally, this region of DNA has 28 to 35 CAGs repeated, interrupted by a sequence that codes for two other amino acids. In patients with SCA1, a mutation resulted in loss of this nonglutamine interruption and more than 35 uninterrupted CAG repeats.
The size of the repeat and the age of SCA1 onset were directly correlated—those with the longest repeats had the earliest onset of symptoms. The repeat disorder found in SCA1 is not unique; other well-known neurodegenerative disorders, such as Huntington's disease and amyotrophic lateral sclerosis, also derive from expansions in stretches of repetitive DNA. This discovery now linked ataxia studies to a much larger body of research on these other diseases. Once the mutation responsible for the development of SCA1 was discovered, researchers could generate a genetic test to identify carriers of the disorder so that family members could receive genetic counseling before conceiving children. Before the identification of the mutation, Larry had been tenaciously counseling presymptomatic Schut family members to wait to have children, and now only a handful of family members were potential carriers of SCA1. However, as SCA1 research gained more media attention, many other affected families were identified around the country. Although the development of a genetic test came too late for many of the Schut family members, it was life changing for other families affected by SCA1.
In 1994, the gene harboring the expanded CAG stretch that caused SCA1 was finally sequenced and named ATXN1 (commonly called Ataxin-1). ATXN1 is a large gene and is translated to protein in both the unaffected and disease versions. However, the protein produced in the SCA1 version is slightly different.
We were unsure what the mutant protein did to cause disease once it was in the nucleus. The list of unknowns was getting longer, not shorter.
Researchers knew that the brainstem and cerebellum were the two areas severely affected in SCA1. Within the cerebellum, they discovered that Ataxin-1 protein was highly expressed within Purkinje cells, which are intricate and highly connected nerve cells critical for coordinating body movements. After they receive signals from planning centers of the brain, Purkinje cells sort out those messages and relay them through the spinal cord to muscles, causing movement. The underlying cause of the ataxia was degeneration of the Purkinje cells in SCA1 patients, and the mutation in Ataxin-1 was somehow involved in causing these cells to die.
Identification of the gene and mutation causing this rare form of ataxia was incredibly exciting for the Schut family and other families affected by SCA1, but many questions about the disease remained to be answered.
As medical interest in ataxia grew in the mid-1990s, researchers still had little knowledge about the normal function of the Ataxin-1 gene, let alone how the diseased gene was initiating the neural degeneration that resulted in ataxia and SCA1. To find answers to such challenging questions about the disease, the Orr lab turned to animal models. Our team identified a related gene in mice that we named Sca1.
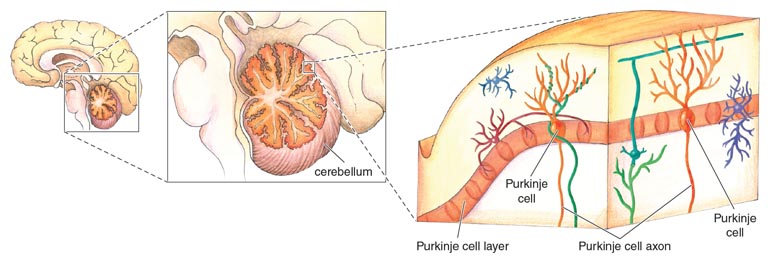
Illustration by Emma Skurnick.
Mouse Sca1 and human ATXN1 are strikingly similar; however, the mouse gene contains only two CAG repeats. To mimic the human disease in the mouse model, we created a transgenic mouse that expressed the human ATXN1 gene with 82 CAG repeats. Eighty-two was the highest number of repeats observed in a human SCA1 patient, and this individual experienced disease onset when younger than five years old. As seen in human SCA1 patients, transgenic mice with 82 CAG repeats had progressive ataxia and degeneration of the Purkinje cells.
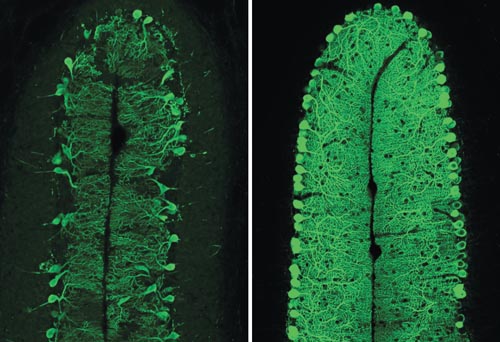
Image courtesy of the Orr lab.
The mice proved to be an excellent model system to study the disease, and they allowed us a closer look at what might cause cell death. We observed clumps of proteins called aggregates forming within the Purkinje cells of diseased animals. It made sense to conclude that the aggregates forming in the nucleus were toxic and resulted in cell death. However, questions remained about what, if anything, these aggregates were doing. Were the aggregates causing the disease, or were they a result of it? Was the expanded Ataxin-1 protein clumping together to form aggregates, or was another protein?
To get some answers to these questions, in 1998 we generated another transgenic mouse model that expressed the expanded human ATXN1 gene, but with an added mutation that prevented it from interacting with itself and forming aggregates. We discovered that mice with this mutation still developed ataxia and underwent Purkinje cell death, but they did not form aggregates in their Purkinje cells. The finding showed that aggregates were not the cause of cell death and disease.
That same year, we also created a mouse completely lacking the normal Sca1 gene to study loss of Ataxin-1 function. These mice did not develop ataxia or have any Purkinje cell death, which indicated that the mutation in Ataxin-1 was not causing a nonfunctional protein. However, mutant Ataxin-1 was doing something to instigate the disease.
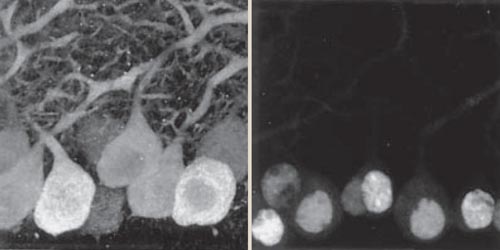
Image from Klement, I. A., et al. Cell 95:41.
Ataxin-1 is normally located in both the cytoplasm and the nucleus of Purkinje cells. We next discovered that Ataxin-1 had to enter the cell nucleus to cause disease. Once it entered the nucleus, the expanded Ataxin-1 protein had difficulty transporting back out. If we kept Ataxin-1 out of the nucleus, we could potentially halt progression of the disease. This seemed like a simple task, but again we faced another hurdle: We did not know what controlled Ataxin-1's entrance into the nucleus. We knew there could be other genes or factors involved in regulating Ataxin-1. And we were unsure what the mutant protein did to cause disease once it was in the nucleus. The list of unknowns was getting longer, not shorter.
Researchers quickly appreciated the complexity of the disorder and our limited understanding of the disease. Undeterred by the challenge, the field of ataxia research continued growing quickly. Each research team tackled different critical questions about the disease. We are still asking these and many other questions. We are not certain whether the mutant Ataxin-1 protein retains much of its normal function, or whether it develops entirely new functions. The complexity of Ataxin-1 biology makes development of a cure slow going.
Our lab, along with Zohgbi's lab, is carrying out high-throughput screens, a current method for rapidly identifying important compounds in a biochemical pathway, to find molecules that reduce the levels of Ataxin-1 protein. We also are investigating the effects of modifying the Ataxin-1 protein and the implications for its biological activity. For example, like many enzymes, Ataxin-1 is turned "on" or "off" depending on whether a phosphate group is added to it or not, which regulates its interaction with other molecules that block Ataxin-1 from entering the nucleus. By suppressing the activities of proteins responsible for adding a phosphate group to Ataxin-1, we may be able to improve the condition of the cerebellar Purkinje cells and reduce the severity of ataxia. We are conducting these studies in mouse models, and we still have much to accomplish before a potential treatment for SCA1 could go through clinical trials in humans.
Understanding the biology behind SCA1 will spur drug discovery and progress in research of other related neurodegenerative disorders. Therefore, the fight of one family struggling with this rare disorder is also affecting other areas of disease research.
Because the family has received genetic counseling, Schut family reunions are no longer opportunities to collect blood samples for genetic testing; instead, they can focus on food and socializing, just as other families do. Only one family member is currently affected by the disease, and another is untested for ataxia and under the age of eighteen. Larry Schut continues to work as a neurologist and to have an interest in a variety of newly identified forms of spinocerebellar ataxia. The family remains active with the National Ataxia Foundation, which in contrast to its humble beginnings, today spends over $1 million annually to support ataxia research.
Despite all these advances, there are currently no clinical-stage treatments for hereditary and sporadic ataxias, and there is still only limited public awareness about the disease. Even in families where hereditary ataxia has presented in multiple generations, patients may struggle for an accurate diagnosis because the disease can be mistaken for other movement disorders, such as multiple sclerosis or amyotrophic lateral sclerosis. Educating the public and providing support for patients affected by ataxia is a major goal of the National Ataxia Foundation.
SCA1 remains at large in other families across the United States, including hundreds of at-risk individuals who may pass the disorder along to their children. Thankfully, the fate for these families is much better than what was faced a century ago, or even 50 years ago. Families have a genetic test for the disease, and government assistance provides help for those affected by this debilitating disorder. Researchers continue to progress toward the next breakthrough to understanding this disease. Decades of dedication from patients, families, and the research community brought scientists closer to understanding hereditary forms of ataxia and identifying a treatment for the disease.
One of us (Nissa Mollema) began researching ataxia after finishing her Ph.D. in 2010 and joining the Orr lab shortly thereafter. Only a few months into the job, a conversation with her mother-in-law led her to discover that perhaps something beyond scientific interest had directed her to find a place in ataxia research. Mollema learned that her husband's great-grandmother had seven siblings affected by SCA1, and that one of these siblings was Henry and John Schut's father.
Reflecting on the early days of ataxia research, Mollema was struck by how the discoveries that paved the way for her research were driven by a simple desire to improve life for the next generation and to prevent the suffering the current generation faced. Questions about SCA1 and a host of related neurodegenerative diseases remain, but collaboration and determination continue to advance our understanding of the larger scientific puzzle.
Click "American Scientist" to access home page

American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.