Mitochondrial Death Channels
By Keith A. Webster
In heart attacks, cells die if they aren't perfused with fresh oxygen—and kill themselves if they are. Understanding cell suicide may greatly improve outcomes.
In heart attacks, cells die if they aren't perfused with fresh oxygen—and kill themselves if they are. Understanding cell suicide may greatly improve outcomes.

DOI: 10.1511/2009.80.384
Coronary artery disease is the leading cause of morbidity and mortality in North America and Europe. More than 12 million people in the United States have coronary artery disease, and more than 7 million have had a myocardial infarction (heart attack). Chronic stable angina (chest pain) is the initial manifestation of coronary artery disease in approximately half of all presenting patients, and about 16.5 million Americans (more than 5 percent) currently have stable angina.
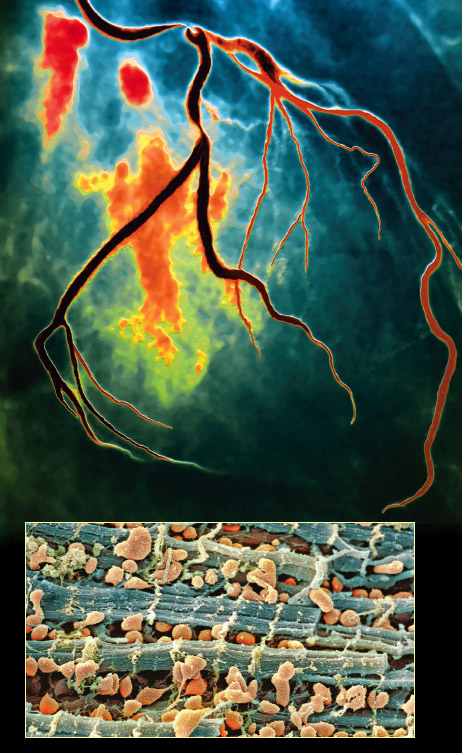
Top image by Zephyr/Photo Researchers, Inc.; bottom image by Steve Gschmeissner/Photo Researchers, Inc.
Surgical interventions to prevent or treat acute myocardial infarction are increasing: More than 1.7 million coronary angioplasty and coronary artery bypass procedures were performed in 2006 in the United States. A major goal of clinical cardiac research is to establish treatment criteria that will limit lethal damage to the heart caused by acute myocardial infarction. Research has focused on the multiple stresses that are unleashed by a heart attack and the effects of these stresses on the intracellular structure and function of cardiac muscle.
The mitochondrion, powerhouse of the cell, is the central player in defining the outcome of heart attacks. Mitochondria contain cellular poisons that are normally sequestered in inactive form, but when unleashed and activated they enforce cell suicide. These suicide regulators are released from mitochondria through mitochondrial death channels. Understanding how the death channels work may hold the key to new treatments that could dramatically reduce myocardial injury and improve the outcome for patients who experience acute myocardial infarction.
A heart attack can affect 50 percent or more of the myocardial left ventricle (which takes in freshly oxygenated blood), causing massive tissue loss and scarring, which is known as infarction. Heart attacks begin with thrombosis; a blood clot wedged in a coronary artery causes reduced blood flow to downstream tissue (ischemia). The cardiac muscle becomes hypoxic (short of oxygen) and acidotic, and the energy level falls because the lack of oxygen interrupts mitochondrial metabolism. Cadiac tissue severely affected by ischemia may cease to contract. Ischemia must be relieved in a timely manner or all of the tissue downstream of the blood clot will die. Relief occurs when the flow of oxygenated blood to the tissue recommences, a process known as reperfusion. The amount of tissue salvaged by reperfusion is determined by the amount of time between the onset of ischemia and removal of the clot.
Clot removal usually involves angioplasty. A flexible needle catheter is inserted into the obstructed coronary artery and a balloon is inflated in the region of the obstruction, compressing the clot against the vessel wall. A stent made of stainless steel—or, lately, high-tech shape-memory alloys—is usually deployed by the catheter in the same region to trap the compressed plaque in place and keep the vessel lumen open when the catheter is removed. When reperfusion delivers oxygen back to the tissue, mitochondria become reenergized and contractions resume. If the ischemic period is short, damage to the heart may be minimal at the onset of reperfusion. However, lethal injury spreads insidiously across the formerly ischemic region over the hours, days and sometimes weeks after reperfusion. This damage, known as reperfusion injury, was first described about 20 years ago. For many heart attack victims, it is the greatest threat to survival.
As reperfusion injury develops, heart cells are forced into a wave of suicide known as apoptosis, or programmed cell death. The stimuli for this are a combination of the reoxygenation component of reperfusion and an imbalance of calcium ions and protons that develops during ischemia and which is exacerbated by reperfusion. The targets for both of these stimuli are the mitochondria. Reperfusion injury begins when mitochondrial death channels open and release the suicide activators. We are only now beginning to fully understand what causes the death channels to open and how the suicide process works. With a fuller understanding, it may be possible to design pharmacological procedures that prevent some of the tissue damage (and deaths) caused by heart attacks.
Coronary artery occlusion usually results from a combination of blood vessel narrowing due to atherosclerosis and a thrombus (blood clot) that wedges in the bottlenecked artery and stops blood flow. The impact on the tissue downstream of the occlusion is immediate, including severe chest pain caused by ischemia and disrupted energy metabolism in cardiac muscle. Ischemic tissue becomes rapidly hypoxic as mitochondria consume all available oxygen to generate ATP, the high-energy molecule that drives most energy-requiring reactions of the cell. When the oxygen is depleted, heart muscle cells (cardiac myocytes) in the ischemic region begin to generate energy anaerobically (without oxygen) by glycolysis. Glycolysis is an ancient energy-generating system in which glycogen and glucose are broken down enzymatically in stages to form the end product lactic acid. Two moles of ATP and two moles of lactic acid are produced for every mole of glucose used. The glycolytic ATP helps sustain life in the ischemic myocardium, but the excess lactic acid cannot be cleared because there is no blood flow to carry it away to the liver, where under normal conditions it would be further metabolized.

Stephanie Freese
Model systems of heart attack reveal that the pH of ischemic myocardial tissue falls from the physiological value of 7.4 to less than 6.0 within 30 minutes of the coronary artery occlusion. Eventually glycolysis is also inhibited, due to the rise in acidity and depletion of locally available glucose. When this happens, there is no alternative energy supply for the formation of ATP. A precipitous drop in ATP results in the interruption of a variety of biochemical pathways, the collapse of essential ion gradients across membranes that are maintained at the expense of ATP, and finally tissue death, called necrosis. Although necrotic death of some myocardial tissue is inevitable during the ischemic period of a heart attack, the extent of it may be quite small. Most heart attack victims in the United States and Europe are hospitalized and have the blood clot removed within three to four hours of the onset of symptoms. In these cases ischemia may cause minimal tissue loss. Paradoxically, however, massive amounts of tissue may be lost as a result of reperfusion.
Reperfusion injury begins immediately when fresh blood returns to the heart, delivering oxygen that allows the operation of the central energy-extracting pathway of mitochondria, the electron-transport chain.
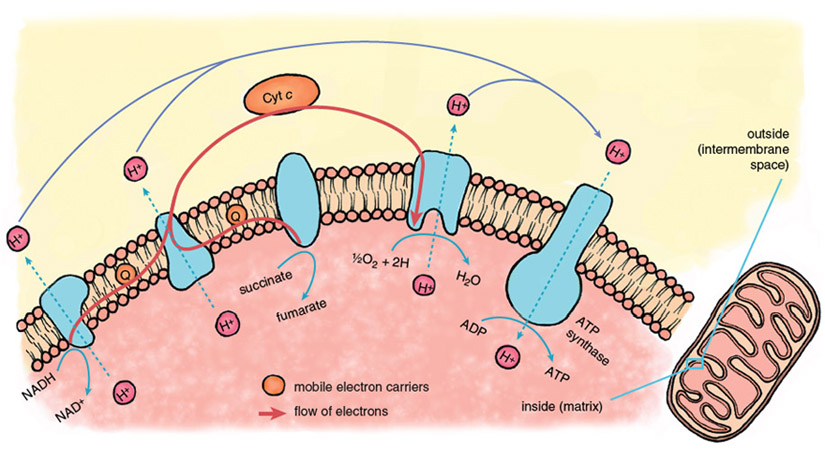
Stephanie Freese
The mitochondrial electron-transport chain is a series of protein-metal complexes embedded in the mitochondrial inner membrane that have progressively higher reduction potentials (affinity for electrons). Electrons derived from the metabolic breakdown of fuel molecules are ferried by mobile electron carriers to specific acceptor complexes, and energy is released as the electrons move down the chain, eventually reaching oxygen, the final electron acceptor. Energy recovered from electron transport is used to expel protons (hydrogen ions, H+) from the mitochondrial interior, creating a gradient in the concentration of protons across the mitochondrial inner membrane.
Considerable potential energy is stored when ion gradients across membranes are created, arising from the imbalances of charge and osmotic potential across the membrane. The energy contained in the mitochondrial proton gradient is transformed into the chemical energy of ATP by another large protein complex in the mitochondrial inner membrane, the proton-translocating ATP synthase. This enzyme complex couples the energy-releasing return flow of protons into the mitochondrial interior with a reaction that forms the high-energy phosphoryl bonds of ATP.
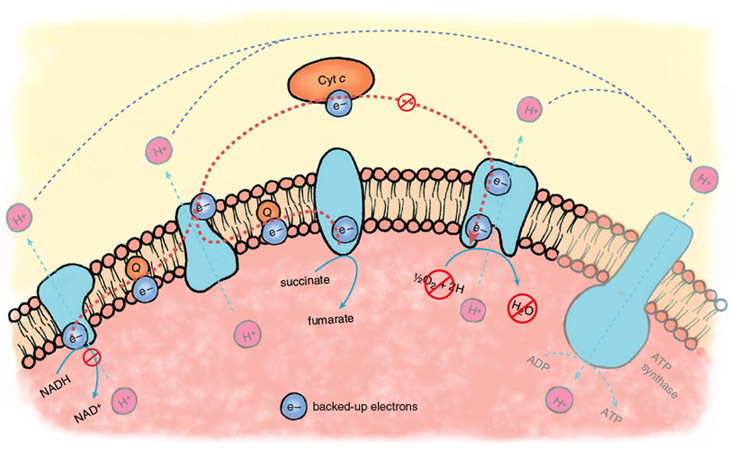
Stephanie Freese
During the ischemic phase of a heart attack, electron transport backs up because of the lack of oxygen as a final acceptor of electrons. The electron carriers of the transport chain, unable to hand off electrons to the next acceptor in line, remain in their reduced state. In this backed-up condition, certain components of the electron-transport chain are primed to generate oxygen free radicals when oxygen returns. There is strong evidence that oxygen radicals play a major role in the initiation of reperfusion injury.
During the normal functioning of the electron-transport chain, a small number of electrons escape and can react with oxygen to form oxygen free radicals or oxidize other cellular components directly. These reactions are mostly neutralized by the cellular anti-oxidant systems—glutathione reductase, superoxide dismutase and catalase, enzymes that are present at high levels in most cells and tissues. During reperfusion, there is a massive increase in the diversion of electrons from the electron-transport chain, such that endogenous antioxidants are overwhelmed, and large amounts of oxygen free radicals escape.
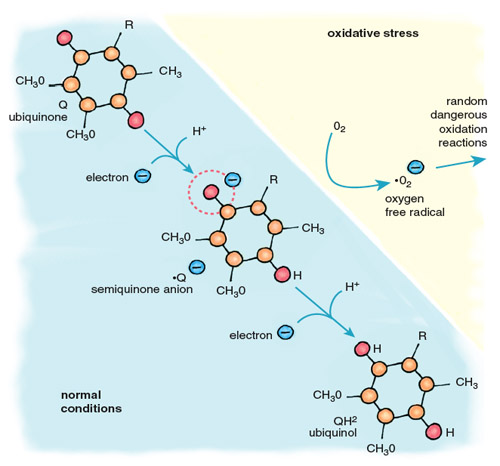
Stephanie Freese
Oxygen free radicals are highly reactive, and those that escape the antioxidant systems immediately attack the nearest cellular components, including proteins, lipids and DNA. The major contributor of electrons for the formation of oxygen free radicals is the mobile electron carrier ubiquinone, also called coenzyme Q (Figure 5). Cycling between two forms, oxidized ubiquinone and reduced ubiquinol, Q accepts electrons from complexes I and II of the electron-transport chain and transfers them to complex III and cytochrome c. When oxygen is absent, there is no final acceptor for electrons after complex IV. Flow through the electron-transport chain is blocked, leaving the carriers in a reduced state, anxious to donate their electrons. When this happens, the ubiquinol pool becomes flooded with electrons, causing an increase in the relative concentration of ubisemiquinone, a reactive free radical form of ubiquinone. When oxygen returns to the mitochondria, as it does when the heart is reperfused after ischemia, ubisemiquinone donates electrons directly to oxygen, generating superoxide anion (•O2–). This diffusible free radical reacts rapidly with neighboring molecules, causing damaging oxidation.
One of the targets of superoxide is the first mitochondrial death channel, known as the mitochondrial permeability transition pore (mPTP), which normally regulates the flow of specific materials between the cytoplasm and the mitochondrial interior. Key components of the mPTP are present in the same membrane system as the electron-transport chain. Oxidation of these mPTP components can cause the pore of the permeability complex to open, allowing the unregulated escape of mitochondrial consitituents and initiating cell death. However, oxidative stress alone does not effectively launch cell death. Another component, calcium, collaborates with oxygen free radicals to open the mPTP.
Calcium plays a pivotal role in the muscle contraction cycle. Systole, the contraction phase of the heartbeat, is initiated by electrical pulses known as action potentials, which stimulate the release of calcium from a membranous compartment called the sarcoplasmic reticulum (SR). The process begins when a small amount of extracellular calcium enters the muscle cell through a pore in the cell membrane called an L-type channel (Figure 6). This calcium interacts with another channel inside the cardiac myocyte called a ryanodine receptor, causing a powerful release of calcium from the sarcoplasmic reticulum such that the intracellular free calcium concentration increases approximately 100-fold. This calcium release triggers muscle contraction.
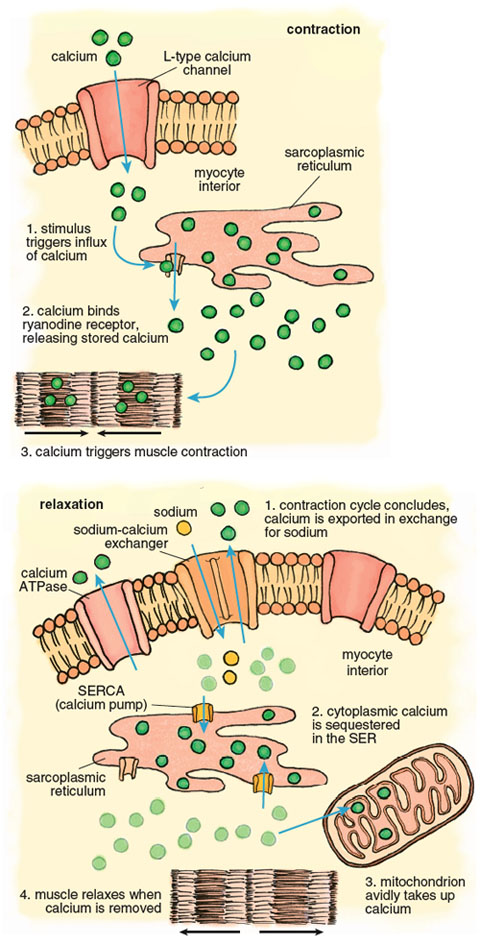
Stephanie Freese
Contraction of the left ventricle pumps oxygenated blood out of the chamber into the peripheral blood vessels. When the left ventricle relaxes, an activity known as diastole, calcium is pumped back into the SR by the SR-calcium ATPase (known as the SERCA channel), and back out of the muscle cell by another calcium ATPase, this one embedded in the cell membrane. As the muscle relaxes, the ventricle chamber expands and oxygenated blood from the pulmonary artery flows in to refill the chamber.
The large calcium fluctuations that drive the contraction-relaxation cycle affect other intracellular components, particularly mitochondria. Mitochondria are calcium sinks. They scavenge calcium via a highly efficient calcium uniporter—a channel that translocates an ion across a membrane without concomitant transport of an oppositely charged ion in the same direction or a similarly charged ion in the opposite direction, which would be necessary to maintain overall charge balance. Counterbalancing of charge during ion transport is a critical feature of normal cell function and post-ischemic recovery. The calcium uniporter transports calcium into the mitochondrial interior (called the matrix) down the electrochemical gradient across the inner membrane. It normally functions in coordination with a sodium-calcium antiporter, with calcium homeostasis maintained by extrusion of calcium in return for sodium. The finely balanced activities of these two channels are disrupted when intracellular calcium increases during both ischemia and reperfusion.
All calcium movements and channel activities must be precisely coordinated so that the concentrations in the different cellular compartments maintain appropriate levels during and between contractions. The resting human heart contracts at about 70 beats per minute, or 4,200 beats per hour. Therefore an imbalance of calcium flux during the contraction cycle is rapidly amplified. In myocardial arrhythmias, including the condition known as atrial fibrillation, erratic calcium movements cause irregular contractile states that can be lethal.
Calcium imbalance develops rapidly during ischemia-reperfusion, with dire consequences. Calcium enters cardiac muscle through L-type channels to initiate contraction and is removed during relaxation by two channels, the calcium ATPase and the sodium-calcium exchanger, both present in the cell membrane of the cardiac myocyte. During ischemia these channels lose coordination, due in large part to the effects of anaerobic glycolysis and acidosis. The rate of glycolysis increases 10- to 20-fold during ischemia, accompanied by high acid production and a rapid decline of pH inside cardiac myocytes. The myocytes respond by activating another channel called a sodium-H+ exchanger that expels H+ in an electroneutral exchange for sodium. This reduces acidosis but increases the sodium concentration inside the myocyte. When the intracellular sodium reaches a certain threshold, it forces the sodium-calcium exchanger to reverse, so that sodium is expelled in exchange for calcium. The net effect is a progressive increase in calcium in the cell that continues for the duration of ischemia.
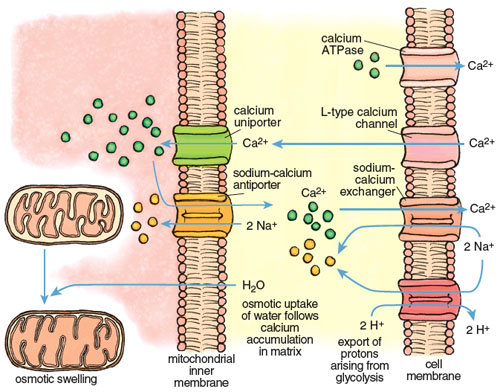
Stephanie Freese
The excess calcium is immediately taken up by the mitochondria through the mitochondrial uniporter. The insatiable uniporter accumulates calcium as long as an electrochemical potential (negative inside) exists across the mitochondrial inner membrane. During ischemia, glycolytic ATP is used to maintain the potential, so calcium flows into the mitochondrion as long as there is glycolytic ATP. As illustrated in Figure 7, calcium accumulation is followed by water intake down an osmotic gradient, causing swelling of the mitochondrial matrix and stretching of the inner membrane. This swelling can open the mPTP and may even burst the outer mitochondrial membrane. Paradoxically, calcium uptake and swelling continue after reperfusion, due to a transient pH imbalance created by fresh blood arriving at the cell. The imbalance occurs because the extracellular acidosis that was built up during ischemia is immediately neutralized, but the cell interior remains transiently acidotic. This causes a transient outside-to-inside proton gradient across the cell membrane that stimulates proton extrusion through the sodium/H+ antiporter and an antiparallel uptake of calcium by the sodium-calcium exchanger. The rise in calcium in the cell cytoplasm drives an increase of calcium inside the mitochondria.
The toll of a heart attack on mitochondria thus includes calcium accumulation and swelling during ischemia and reperfusion, followed by a surge of electrical current through the electron-transport chain during reperfusion, and superoxide production that overwhelms antioxidant defenses. These forces determine whether mitochondria transmit death or survival signals to cardiac muscle cells.
All mammalian cells, including cardiac myocytes, are endowed with a capability to die by voluntary as well as involuntary processes. Involuntary death is usually caused by extreme cellular damage that precludes repair, such as the necrotic death that results from prolonged ischemia with severe energy depletion. Voluntary death is a form of cell suicide. During development it occurs when cells need to be removed and replaced by more specialized cells required for organogenesis. It can also follow damage that is nonlethal but may handicap cellular function. Voluntary cell death is a programmed action that avoids collateral damage. It usually involves maintaining the dying cell with an intact cell membrane until the defective cell is recognized by the immune system as nonnative and safely removed. The most common form of cell suicide is termed apoptosis, from the Greek, apo-, from, and ptosis, falling, referring to leaves falling from trees or petals from flowers. Two forms of programmed death are associated with heart attack: classical apoptosis and programmed necrosis. They are both regulated by the mitochondria and together they are responsible for most of the injury that follows reperfusion. Both death pathways are initiated by opening of the mitochondrial death channels and both may be preventable.
The mitochondrial death channels include the mitochondrial permeability transition pore (mPTP) and the mitochondrial apoptosis channel (mAC). The mPTP has been under intense study for almost half a century, but its composition and mechanism of action are still controversial. Most experts agree that the mPTP is a voltage-dependent channel that is regulated by calcium and oxidative stress. Most but not all agree that three distinct mitochondrial proteins can influence the function of the mPTP. Two of these proteins are normally engaged in transporting metabolites into and out of the mitochondria. The first and outermost component of the mPTP is the voltage-dependent anion channel (VDAC), present in the mitochondrial outer membrane and required for the transport of ATP and ADP between mitochondria and the cytoplasm. The second component is the adenine nucleotide translocator, a transporter protein that exchanges ATP for ADP across the inner mitochondrial membrane, thus ferrying ATP in the matrix to the VDAC for export to the rest of the cell. The third component, cyclophilin D (CypD) ensures the proper folding of newly synthesized mitochondrial proteins and has attracted special interest because of its response to certain drugs. CypD is localized on the matrix side of the inner membrane. Together the three proteins of the mPTP span both mitochondrial membranes and provide a pathway between the matrix and the cell cytoplasm.
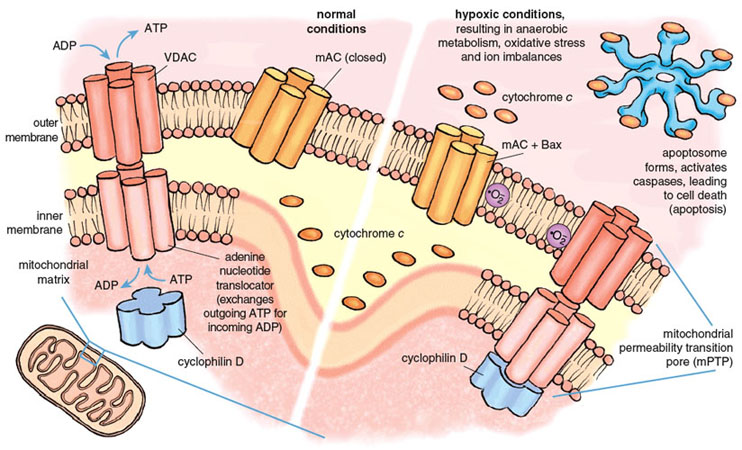
Stephanie Freese
Under physiological conditions, the mPTP is closed to molecules that are not substrates of VDAC or the adenine nucleotide translocator. Conditions prevailing during a severe heart attack cause the mPTP to open, probably during early reperfusion. Opening of the mPTP allows the unregulated transport of materials out of the mitochondrion, and initiates both necrotic and apoptotic death. Death regulated by the mPTP channel causes 50 percent or more of the lethal injury caused by a heart attack.
The second death channel, known as the mAC, was discovered only recently (in 2004). It is responsible for classical apoptosis. Multiple life-threatening stimuli cause the mAC to open, allowing the release of the suicide inducers. The activity of the mAC is under tight control by a family of proteins collectively known as the Bcl-2 family, of which an important member is called Bax. Bcl-2 derives its name from B-cell lymphoma 2, the second member of a family of proteins whose over-expression is linked with lymphoma. The combined actions of the mPTP and the mAC are responsible for most and possibly all of the deaths caused by reperfusion during a heart attack.
The individual roles of the two death channels in promoting infarction have been dissected using specific inhibitors and genetic experiments in which individual genes associated with the channels are deleted, and the effects of the deletion on channel function and programmed death are determined. These studies have been done mainly using mice. The matrix component of the mPTP, CypD, is selectively bound and inhibited by an immunosuppressant drug called cyclosporine A. For many years we have known that treatment of animals with cyclosporine A reduces the damage of acute myocardial infarction. Most researchers in the field thought that this was because cyclosporine A suppressed apoptosis. However, recent studies on mice with homozygous CypD gene deletions (CypD –/–) indicate that this may not be true. CypD –/– mice were found to be resistant to lethal injury caused by a heart attack, but the effect was due to decreased necrosis, not decreased apoptosis.
The studies confirmed previous findings that calcium and oxidative stress caused the mPTP to open, and significantly more calcium and oxygen free radicals were required to open the mPTP when CypD was absent. However, CypD deficiency had no effect on apoptotic cell death when cells were treated with classical inducers of apoptosis. This means that necrosis but not apoptosis requires an intact mPTP and a functional CypD to be activated during a heart attack. It also suggests that this form of necrosis is voluntary (programmed) because it only occurs when CypD is present and can be prevented by genetic deletion of CypD or inhibition of CypD with cyclosporine A. Together these studies support a primary role for the mPTP, regulated by calcium and oxidative stress, in causing necrotic death during a heart attack. They indicate that the mAC can function independently of the mPTP to promote apoptosis, but they do not eliminate the possibility that both channels have roles in the regulation of apoptosis during a heart attack.
Necrosis and apoptosis both contribute to myocardial infarction. A major contribution of apoptosis is indicated by direct measurement of apoptotic markers in tissue of heart attack victims, as well as by the effects of specific inhibitors of apoptosis. The former indicate that between 20 and 50 percent of cardiac myocytes in the area of the left ventricle exposed to ischemia are actively undergoing apoptosis shortly after reperfusion. Consistent with this, selective inhibitors of apoptosis added during a heart attack can reduce infarction by 20 to 50 percent. Numerous studies indicate that opening of the mAC initiates this death pathway.
The mAC launches apoptosis by forming a channel in the outer mitochondrial membrane that allows for the release of cytochrome c, a mobile electron carrier in the electron-transport chain that is associated with the inner membrane and intermembrane space. Cytochrome c interacts with other proteins in the cell cytoplasm to form a death-dealing complex called an apoptosome. The apoptosome in turn mediates the activation of a network of proteases called caspases and DNAses that digest and destroy cellular proteins and DNA. Like the mPTP, the mAC is opened by calcium and oxidative stress during reperfusion, but it appears to be a more sensitive channel that opens at lower levels of stress. Reperfusion has the ingredients to open both death channels, and it seems likely that both death pathways are activated simultaneously. Necrosis caused by the opening of mPTP may be responsible for a major part of the early infarction caused by the heart attack, whereas mAC-mediated apoptosis may contribute more to the extended infarction that develops over time after reperfusion. The pathways leading to activation of the death channels are linked; it has been shown that Bax, the activator of the mAC complex, can interact directly with the sarcoplasmic reticulum, causing the release of calcium that is taken up by mitochondria and that may contribute to opening of the mPTP. There is also compelling evidence that both pathways are regulated by other Bcl-2 proteins.
An underlying feature of the death channels is that they are not inevitable. Observations that CypD- and Bax-deficient mice are significantly protected against the damage of heart attacks suggest that the damage can be largely prevented by blocking the death channels. Indeed, preclinical studies have confirmed that a number of pharmacological agents mitigate the opening of the mitochondrial death channels and can reduce tissue damage from heart attack or angioplasty, with potentially dramatic decreases in infarction and mortality. Along with cyclosporine A, currently approved as an immunosuppressant and widely used during organ transplant procedures, one of the most extensively tested and potent agents is low-dose sildenafil citrate, an agent already approved for erectile dysfunction and marketed as Viagra. It may not be long before an older individual, feeling unwell before bed, perhaps having barely worrisome chest pains, will recall a consultation in the doctor’s office and reach for cyclosporine A, against the chance that angina is about to become something more urgent. The threat can be minimized if medical care is near at hand, and if the mitochondrial death channels can be coaxed into remaining closed.
Click "American Scientist" to access home page

American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.