Metformin: Out of Backwaters and into the Mainstream
By Philip A. Rea, Anderson Y. Tien
This ubiquitous diabetes drug took a convoluted route to become the standard of care, and is still finding new uses.
This ubiquitous diabetes drug took a convoluted route to become the standard of care, and is still finding new uses.

Metformin has come to prominence despite a checkered history. Its story is one of delays, uncertainties, and dead ends, as well as fortuitous accidents. It is a drug, born of folklore, with reasonably well-established clinical benefits but whose precise mechanism of action has resisted definition. Yet, metformin is the current standard of care for the treatment of one of the most common chronic conditions in the modern world—type 2 diabetes—and its chemical structure is remarkably simple by comparison with that of many other drugs.

Antonio Romero/Science Source
Gillian Shenfield, writing in the Australian Prescriber in 2013, summed up metformin’s path well:
Clinical trials of new drugs may overstate efficacy and not identify adverse effects. It is therefore unusual for the passage of time to reveal that a drug is less toxic, has greater efficacy, and a wider range of uses than first claimed. For decades metformin was misunderstood, vilified, and banned in many countries, but it is now one of the most prescribed drugs in the world. In 2010 there were more than 100 million prescriptions worldwide for metformin, alone and in combination tablets.
Of the many baffling aspects of metformin is the haphazard way in which it first got noticed and came to assume the standing it now has at very disparate times—separated by decades—in different parts of the developed world. Unforeseen too is the extent to which its applications have since expanded from the treatment of type 2 diabetes to the treatment of prediabetes and polycystic ovary syndrome (PCOS), and even cancer.
Several factors influenced metformin’s uncertain rise to fame, most notably the landmark discovery of insulin in the early 1920s; the disruptive effects of the First and Second World Wars on research efforts; and the aftershock of fear among American patients when phenformin, a drug in the same class as metformin, was found to be harmful after it had been marketed and prescribed extensively throughout the 1960s and 1970s.
To get a sense of the scale of metformin’s impact, it is necessary to understand what it treats. Diabetes mellitus, or simply diabetes for short, comes in two major forms: type 1 and type 2, which account for 5-10 percent and 90-95 percent of cases, respectively.
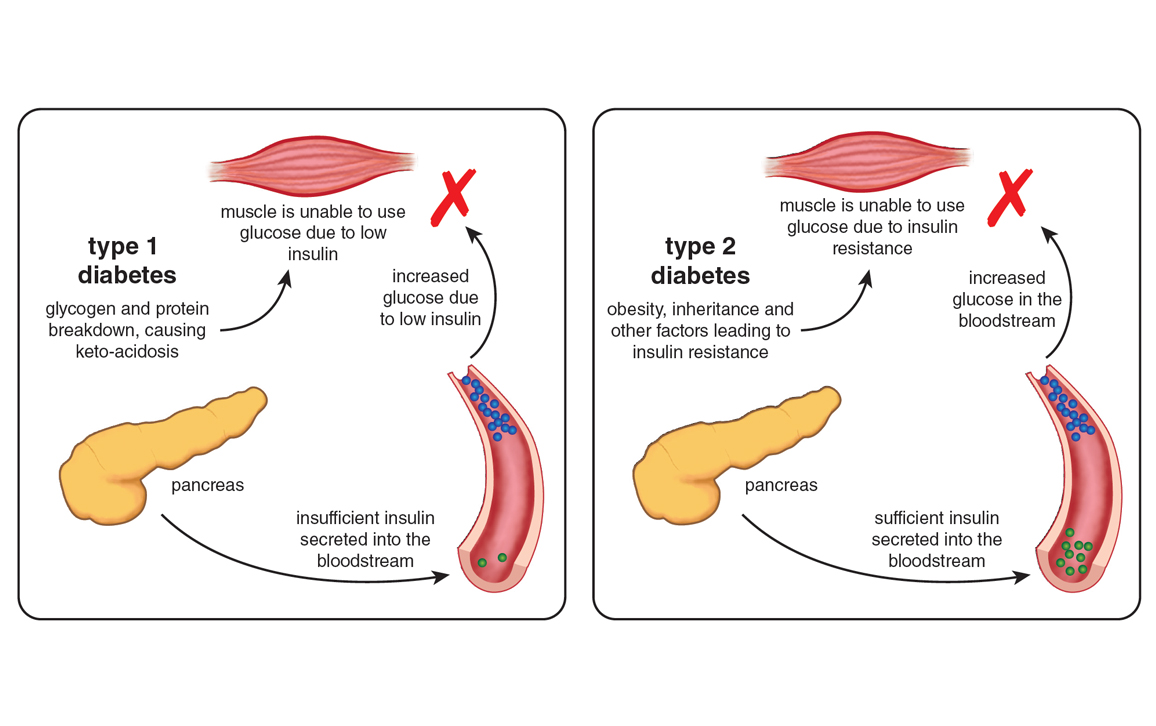
Illustration by Barbara Aulicino.
Type 1 diabetes, otherwise known as juvenile-onset or insulin-dependent diabetes, first shows itself early in life, with the peak age for diagnosis in the mid-teens. Caused by markedly impaired insulin production because of autoimmune destruction of the insulin-producing beta cells of the pancreas, type 1 diabetes is usually fatal unless treated with subcutaneous insulin injections.
Type 2 diabetes, or adult-onset or non-insulin-dependent diabetes, is usually diagnosed later in life than type 1, typically in older, overweight or obese individuals. It begins with insulin resistance, a condition in which muscle, liver, and fat tissues exhibit a diminished response to insulin. Although type 2 diabetics produce insulin, it doesn’t trigger the absorption of glucose from the bloodstream as well as it does in nondiabetic individuals. This deficiency means that a vicious cycle of elevated blood glucose levels (hyperglycemia) and increased insulin production is established, which can eventually exhaust the insulin secretory capacity of the pancreas.
Diabetes is the seventh-leading cause of death in the United States. Some 29.1 million people, or 9.3 percent of the population, have diabetes, of which about 8.1 million have yet to be diagnosed. Moreover, it is estimated that about 37 percent of U.S. adults aged 20 years or older and 51 percent of adults aged 65 years or older—roughly 86 million Americans—have prediabetes. Although a prediabetic has blood glucose levels higher than normal but not high enough to qualify as a true type 2 diabetic, he or she is likely to succumb to the disease within 10 years or so and may already have incurred some of the cardiovascular complications associated with the condition.
Diabetes and prediabetes come at great cost, from not only a humanitarian but also an economic standpoint. As of 2012, the total direct medical costs and indirect costs (measured as disability, work loss, and premature death) in the United States were $176 billion and $69 billion, respectively, giving a total cost of $245 billion. This cost is aggravated by the fact that the incidence of type 2 diabetes, which is associated with a 10-year decrease in life expectancy, is on the rise. In 2010, approximately 285 million people were diagnosed with the disease worldwide compared to 30 million in 1985, and the World Health Organization predicts that as many as 366 million individuals will be affected by 2030.
In 2010, about 285 million people were diagnosed with diabetes. The World Health Organization predicts that as many as 366 million will be affected by 2030.
Some type 2 diabetics can manage their blood glucose levels through dietary changes and exercise alone, but if the levels are not adequately controlled in this way, oral diabetes medications or insulin injections are required. Of the various oral antidiabetic treatments available, metformin is considered to be the gold standard: It is often the first to be prescribed, along with lifestyle changes, when this condition is diagnosed. Metformin acts to diminish the liver’s production of glucose (gluconeogenesis) and improve glucose uptake from the bloodstream so as to offset the effects of insulin resistance. Metformin is particularly effective in the treatment of type 2 diabetes because its effects, unlike those of some of the other oral antidiabetic drugs and insulin, are not associated with a gain in body weight or an increased risk of hypoglycemia (low blood sugar levels).
Metformin’s roots—or more correctly, the origins of its antecedents—can be traced back to the use of the wild legume Galega officinalis as an herbal medicine in medieval Europe. A perennial herb with white, blue, or purple flowers, it is found in most temperate regions. Variously known by the common names goat’s rue, French lilac, Italian fitch, professor-weed, Spanish sainfoin, or false indigo, the aboveground parts of this plant have been used to treat, among other things, the plague, snake bites, and St. Vitus dance (Sydenham’s chorea, a manifestation of acute rheumatic fever).
The first-known written account of the use of goat’s rue for the treatment of diabetes is in Nicholas Culpeper’s Complete Herbal, first published in the mid-17th century, when English physicians were starting to take the disease seriously. From that time until the 1930s, particularly in France, accounts of the use of G. officinalis extracts for the treatment of diabetes were numerous. In fact, it’s still out there. An Internet search for goat’s rue will return more than 90,000 hits, most of which are sites selling goat’s rue preparations for the treatment of a variety of ailments, including high blood glucose. The plant, however, is designated as a class A poisonous plant in 35 states in the United States and is listed in the U.S. Food and Drug Administration (FDA) Poisonous Plant database.
The first-known written account of the use of goat’s rue for the treatment of diabetes is in Nicholas Culpeper’s Complete Herbal, first published in the mid-17th century, when English physicians were starting to take the disease seriously. From that time until the 1930s, particularly in France, accounts of the use of G. officinalis extracts for the treatment of diabetes were numerous. In fact, it’s still out there. An Internet search for goat’s rue will return more than 90,000 hits, most of which are sites selling goat’s rue preparations for the treatment of a variety of ailments, including high blood glucose. The plant, however, is designated as a class A poisonous plant in 35 states in the United States and is listed in the U.S. Food and Drug Administration (FDA) Poisonous Plant database.
The chain of events that eventually spawned metformin as an oral antihyperglycemic drug did not come from diabetes research but instead from investigations of the parathyroid glands. Around 1915, researchers observed that surgical removal of these structures found in the neck is accompanied by increased excretion of guanidine, another substance in the same class as metformin, called the biguanides. Guanidine was known to induce convulsions similar to those associated with the muscle spasms that accompanied loss of parathyroid function when injected into laboratory animals. C. K. Watanabe, who did his seminal experiments at Yale University, speculated (wrongly, but quite reasonably in terms of what was known at the time) that the cause of such spasms, called tetany, in hypothyroidism is the production of “relatively enormous quantities of guanidine bases in the body as a result of the impairment of parathyroid function.” He then speculated that “if these guanidine bases are the sole source of the symptoms of tetany it seems interesting to inquire whether there is a correlation between the hypoglycemia produced by the parathyroidectomy and the guanidine content of the blood.” This connection is what he examined. In experiments on rabbits published in 1918, Watanabe established that the injection of guanidine elicits hypoglycemia.
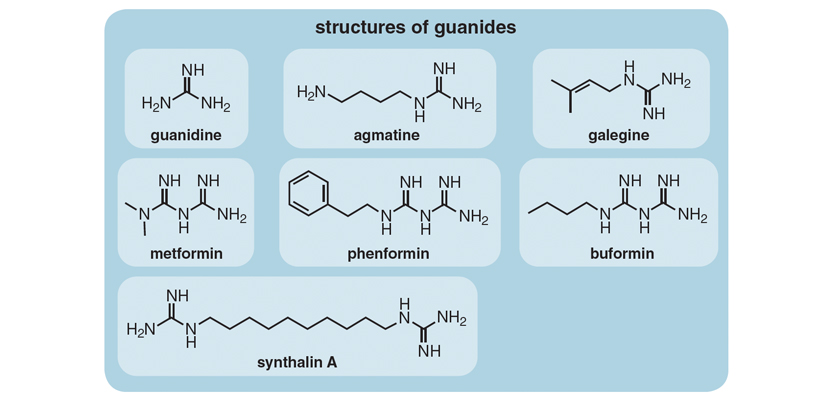
Unfortunately, guanidine was too toxic to be of clinical value. Watanabe’s discovery, however, together with the realization that extracts of goat’s rue are also a rich source of this compound, was to intersect productively with the findings of French pharmacist and plant biochemist Georges Tanret. In 1914, while at the University of Paris, Tanret isolated an alkaloid called galegine from seeds of G. officinalis. These advances were critical, because together they closed the loop between what was already known of the antidiabetic effects of goat’s rue and the chemical identity of a guanidine derivative from this source that was to prove less toxic.
Interestingly, Tanret initially misidentified galegine’s structure, then World War I interrupted work on the substance. So it wasn’t properly identified and synthesized by other groups until 1925. It was not until 1927, some 13 years after its initial isolation, that Tanret and his colleagues published a detailed account of galegine use in rabbits and dogs that categorically demonstrated its hypoglycemic effects. While these trials were being conducted in Paris, Helmut Müller and Helmuth Reinwein in Munich, who were brave enough to use themselves as the very first human subjects before recruiting others, took this research to the next level—the clinic. In work published the same year, the Germans demonstrated a marked hypoglycemic effect of orally administered galegine in hyperglycemic diabetic patients.
Another potentially momentous line of attack on diabetes coincided with the research on galegine but ultimately was less influential, though perhaps for arbitrary reasons. Erich Frank of Breslau, Poland (now Germany), found that another natural plant product, agmatine, a derivative of guanidine, is also a hypoglycemic agent.
Though agmatine did not make it to the clinic, a byproduct of its synthesis did. In the course of their attempts to synthesize agmatine with an eye to commercialization, Schering-Kahlbaum AG of Berlin, the chemical company with whom Frank was collaborating, accidentally synthesized a related compound, decamethylene diguanide. Dubbed Synthalin, this compound was found to be better tolerated and more effective than either galegine or agmatine. Indeed, the results obtained with Synthalin were considered sufficiently encouraging for Schering AG to market it for the treatment of mild cases of type 2 diabetes—though not for long, however, because adverse digestive, hepatic, and renal complications, that were at least construed to be drug-related, soon appeared.
Then Synthalin B (dodecamethylene diguanide), with a slightly longer alkyl chain than its antecedent (which was rebranded Synthalin A), was introduced, although it too was eventually to prove—or perhaps more correctly, to be considered—not safe enough for general acceptance. With the exception of Germany and a few other clinical centers scattered around Europe, Synthalin B use was discontinued in most countries in the 1930s.
Illustration by Barbara Aulicino.
That said, long after the fact, all was not perhaps as it seemed in the ill-fated story of Synthalin B. Jean Sterne, the investigator who decades later was to champion metformin, was more guarded than most in casting aspersions on the Synthalins. He wrote in 1969 that in his sense of things, the “complications earlier ascribed to Synthalin are not particularly convincing, at least as far as its administration to humans is concerned…[in that] many of the side-effects were coincidental and many were due to the presence of viral hepatitis.” Other key players echoed this assessment, conceding that the complications, which at the time were interpreted as a causal relationship between Synthalin administration and hepatic and renal dysfunction, were as likely to represent symptoms of long-standing diabetes (which was not well understood at the time) as to be an adverse reaction to the drug. This admission leaves open the possibility that if more was known of the etiology of type 2 diabetes in the Synthalin era, these agents might have better stood the test of time to gain the widespread use that metformin now has.
Following the Great War, the next big obstacle in the metformin story was a far more positive development for the world at large. The 1921 discovery of insulin by Frederick G. Banting, Charles Best, James Bertram Collip, and John Macleod of the University of Toronto put the kibosh on research into the use of biguanides and other oral antihyperglycemic drugs for the treatment of type 2 diabetes for many years.
Within two years of insulin's discovery, it was brought to market, but its development put the kibosh on research into oral diabetes drugs for many years.
Staggering as it may seem compared to the modern rigors of drug approval, within two years of insulin’s discovery, in 1923—the same year in which Banting and Macleod were awarded the Nobel Prize in Physiology and Medicine for the discovery—Eli Lilly & Company Biological Laboratories, in Greenfield, Indiana, in collaboration with Connaught Laboratories in Toronto, had scaled up the production of insulin to the extent that the company could bring Iletin (the trade name for bovine insulin) to market in the United States. The European market soon followed.
War—the Second World War this time—was again to play its part but in a different way: by rejuvenating, rather than stalling, interest in guanides, biguanides in particular. The Axis countries had blockaded the main production sites of quinine, the standard drug for treating and preventing malaria, so the British assembled a team of chemists and biologists to come up with an alternative. They developed the synthesis of Paludrine in 1945 at Imperial Chemical Industries Ltd. in Blackley, Manchester. What is striking about this compound, which is still used today as an effective antimalarial, is that the right-hand portion of the molecule is a biguanide moiety that bears a close resemblance to the goat’s rue compound, galegine. This discovery did not escape the attention of other investigators. For instance, two years later, K. K. Chen and Robert Anderson at the Lilly Research Laboratories in Indianapolis, while examining the toxicity of Paludrine in animals, discovered that it caused a slight decrease in blood glucose levels.
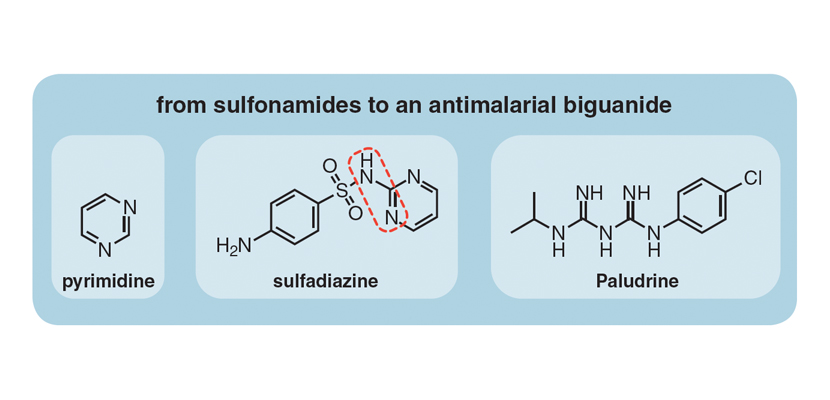
It is unknown for sure if the investigations made by Imperial Chemicals in the United Kingdom and Lilly in United States prompted what happened next. But in the late 1940s in the Philippines, where malaria was endemic, Eusebio Garcia, an infectious diseases physician, was treating patients who had malaria and had also contracted influenza. Historical accounts are vague as to how he acquired what he called a “new synthetic analgesic and anti-flu drug” that he aptly named Flumamine, but what he had in his hands was none other than what we now know as metformin.
Garcia went no further than simply stating that, “The anti-flu potency of ‘Flumamine,’ chemically known as N1-dimethylamino-guanidylbiguanide hydrochloride, was first noted incidentally during the course of biological tests of a number of biguanide derivatives on malaria patients.” He then specified, “Since the polymethylene diguanides have the unusual physiological property of lowering the blood sugar level, it is possible that Flumamine has the same pharmacology”; that is, “if it can lower the blood sugar level to the minimum physiological limit, it can destroy the parasite indirectly by attrition.”
Garcia went no further than simply stating that, “The anti-flu potency of ‘Flumamine,’ chemically known as N1-dimethylamino-guanidylbiguanide hydrochloride, was first noted incidentally during the course of biological tests of a number of biguanide derivatives on malaria patients.” He then specified, “Since the polymethylene diguanides have the unusual physiological property of lowering the blood sugar level, it is possible that Flumamine has the same pharmacology”; that is, “if it can lower the blood sugar level to the minimum physiological limit, it can destroy the parasite indirectly by attrition.”
What makes this leap all the more bewildering is that he provided no experimental evidence for this speculation; no blood glucose levels were reported for his patients. Yet this was to prove a turning point in the metformin story: Sterne, who had added metformin to the diabetes pharmacopoeia, said in an interview toward the end of his life in 1996 that Garcia’s seven-page article in the Journal of the Philippine Medical Association was what got him started on this particular line of investigation.
Sterne trained in diabetology in Paris under Francis Rathery at Hôpital de la Pitié, where he first investigated galegine before moving just a few miles to Hôpital Laennec, while he also held a position with Aron Laboratories, a small pharmaceutical company.
In the 1950s while at Laennec and Aron, Sterne, in collaboration with Denise Duval, conducted studies of the antidiabetic properties of several biguanides, of which one, metformin, was carried forward for clinical research and development. The trade name Glucophage (for “glucose eater”) was first coined for this biguanide in a modest two-page paper the group published in a relatively obscure Moroccan medical journal, Maroc Médical.
There was no disguising Sterne’s cautious optimism when in this paper he wrote, in translation: “Chronic toxicity is virtually nil. Rabbits, rats, and dogs treated with the product over six months showed no deterioration in hepatic function […] [and yet] it has a powerful hypoglycaemic effect […] one can bring the blood sugar down practically as low as one wants.”
On the strength of these preclinical trials, Sterne, Duval, and their colleagues became the first to try metformin in humans for the treatment of diabetes, and Sterne was granted the first patent for this drug. This patent was first filed in 1957, and as continuations in 1958 and 1963, to issue in 1965 under the auspices of Aron Laboratories.
On the strength of these preclinical trials, Sterne, Duval, and their colleagues became the first to try metformin in humans for the treatment of diabetes, and Sterne was granted the first patent for this drug. This patent was first filed in 1957, and as continuations in 1958 and 1963, to issue in 1965 under the auspices of Aron Laboratories.
With the benefit of hindsight, it is perhaps noteworthy that Sterne and his colleagues in France, at Sterne’s own admission, were not aware of work being done on two other biguanides, phenformin and buformin, elsewhere in the world. If the French researchers had known of this research and had shifted their attention toward these two biguanides—both of which turned out to have life-threatening side effects—it could well have meant the death knell of biguanides as a drug class.
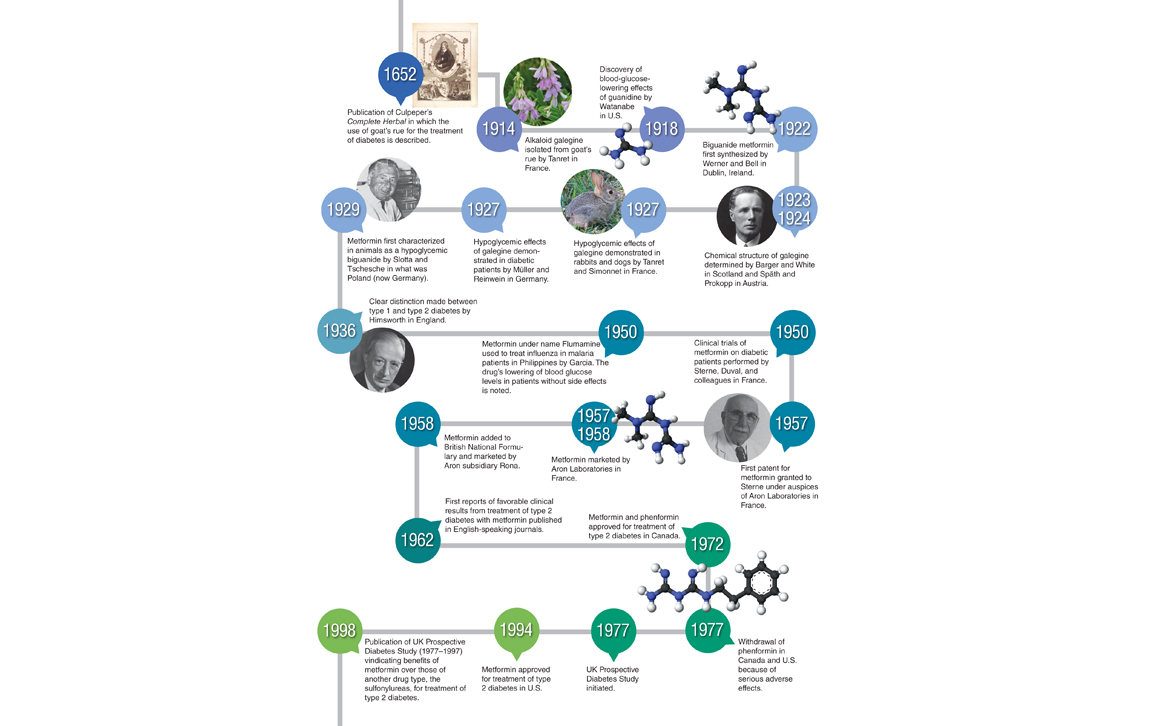
Illustration by Barbara Aulicino.
Metformin’s life as a prescription drug for the treatment of type 2 diabetes began in France, but the possibilities it offered as an oral alternative to insulin were quick to bridge the English Channel. Within one year of its French market entry, metformin was added to the British National Formulary and first sold in the United Kingdom by Rona, a small London-based subsidiary of Aron, in 1958. Modest British clinical trials, published in the British Medical Journal in 1962, conducted on a total of 39 patients over a six-month period, showed that metformin appeared to have considerable potential for the treatment of type 2 diabetics, who represented the majority of the patients in these trials, but not for type 1 diabetics. A similar, smaller-scale study from the University of Edinburgh published in the same year in the British Journal of Clinical Practicecame to much the same conclusion.
These studies and others that were soon to follow made two things apparent to the medical community at large. One is that unlike insulin, which often must be injected several times daily, metformin needs to be administered orally only once or twice a day. The other is that whereas the timing of insulin administration is absolutely critical, the timing for metformin administration is not. If a diabetic misses a meal after injecting himself or herself with insulin, the consequences can be dire—coma brought on by a precipitous decrease in blood glucose. This risk is not the case for metformin, which is not able to drive blood glucose to such dangerously low levels.
Metformin’s triumphant progress around the world was anything but smooth. Its approval for the treatment of type 2 diabetes in Canada in 1972 preceded its approval in the United States by more than 20 years, because the Canadians did something the Americans didn’t—they hedged their bets. The Canadian Diabetic Association (CDA) advisory committee to the health protection branch of the Department of National Health and Welfare recommended not one but two biguanides for the treatment of type 2 diabetes: either phenformin or metformin. This sleight of hand was to prove apt because soon after, when many cases of fatal lactic acidosis (a condition in which the waste products from the anaerobic breakdown of glucose build up too quickly for the body to remove them) among patients taking phenformin were brought to the attention of the CDA advisory committee, it could do what the FDA could not. They simply revised their original endorsement of two biguanides to the endorsement of only one, metformin. This option was not available to Americans in the 1970s.
Metformin's reputation was tarnished because it is in the same drug family as phenformin, which caused fatal side effects in an estimated 4 out of 1,000 users.
That was the first strategic error the American authorities made. The other was their failure to respond promptly to reports of adverse reactions from phenformin. Despite many cases of fatal lactic acidosis associated with phenformin’s use dating from the early 1970s, it was (no two ways about it) aggressively marketed in the United States under the brand name DBI (for Dibotin) by Ciba-Geigy; it managed to survive as a therapeutic until 1977, when pressure from consumer groups was sufficient to compel its withdrawal from the market by the FDA at a time when 250,000 to 385,000 diabetics were still using it. And not a moment too soon. Several years before, in 1973, an FDA medical officer had estimated that 4 out of every 1,000 users of this drug had died of lactic acidosis, which amounted to an annual death toll of around 4,000.
Because it belonged to the same drug family as phenformin, metformin’s reputation was tarnished. It took two decades, until March 1994, for metformin to receive FDA approval. What was by then patently obvious from its worldwide use—in more than 80 countries from which there had never been a need to withdraw it for safety reasons—is that patients on metformin experienced lactic acidosis at only about one-tenth the rate of those on phenformin.
Because it belonged to the same drug family as phenformin, metformin’s reputation was tarnished. It took two decades, until March 1994, for metformin to receive FDA approval. What was by then patently obvious from its worldwide use—in more than 80 countries from which there had never been a need to withdraw it for safety reasons—is that patients on metformin experienced lactic acidosis at only about one-tenth the rate of those on phenformin.
The U.S. patent on Glucophage expired in September 2000, after which many generic substitutes entered the market. As of 1999, sales of Glucophage were at $1.3 billion. Now that low-cost generic formulations are available, metformin ranks as the most widely prescribed antidiabetic drug in the world.
A number of studies would exonerate metformin’s safety and efficacy, but it was not until the landmark UK Prospective Diabetes Study (1977-1997), published in 1998, that metformin was to gain the general level of acceptance it now has. This comprehensive multicenter study, involving more than 4,000 patients who were followed for a little more than 10 years, provided the first clear evidence that overweight and obese patients with type 2 diabetes who take metformin live longer and suffer fewer cardiovascular complications and adverse drug reactions than those who achieve the same blood glucose levels using insulin or other oral diabetes drugs. Similar studies conducted in the United States and Europe, published in 2012 and 2014, further confirmed these findings.
Not only does metformin enhance insulin sensitivity, but it also inhibits intestinal glucose absorption and liver gluconeogenesis to improve glucose tolerance in type 2 diabetics. It does this by lowering basal blood glucose as well as the levels immediately after a meal while at the same time putting the brakes on weight gain and the cardiovascular complications that ordinarily accompany diabetes, all without eliciting hypoglycemic episodes in either type 2 diabetics or nondiabetic subjects.
Many gaps still exist in our understanding of the precise mechanism of action of metformin and other biguanides.
Many gaps still exist in our understanding of the precise mechanism of action of metformin and other biguanides.
It has been known for some time that metformin and biguanides inhibit the activity of mitochondria, the powerhouse organelles of cells that harvest energy from the combustion of foodstuffs to generate the common energy currency of most cells, adenosine triphosphate (ATP). This finding dates back to the pioneering work of Gunnar Hollunger at the University of Lund, Sweden, in the mid-1950s. Metformin and other biguanides compromise mitochondrial respiration to diminish glucose metabolism through the citric acid cycle, so forcing the cells affected to compensate for the loss in energy yield by increasing oxygen-independent glycolysis.
Of the many suspects implicated in connecting metformin’s action on the respiratory chain with its antidiabetic effects, those that feature most highly are the breakdown products of ATP, adenosine 5’-diphophosphate (ADP) and adenosine 5’-monophosphate (AMP).
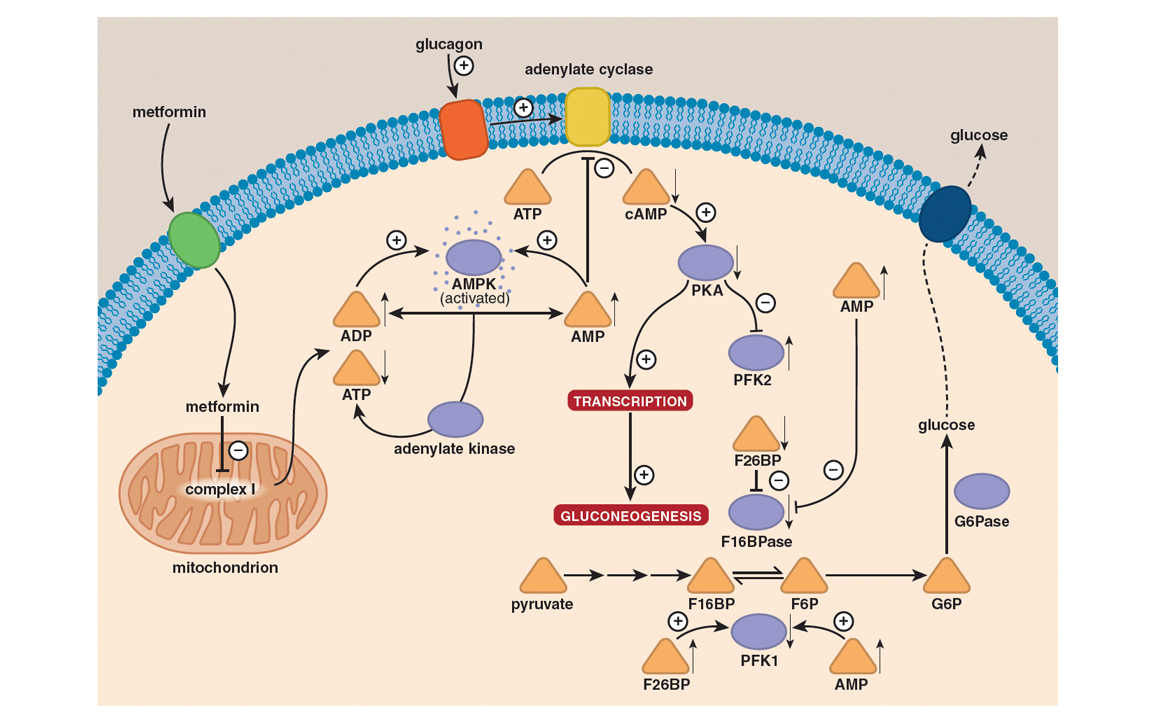
Illustration by Barbara Aulicino.
When attempting to understand what might be going on here, an instructive analogy is that of a rechargeable battery. Whereas the breakdown—catabolism—of foodstuffs such as glucose charges the battery by converting ADP to ATP, most other cellular processes drain the battery by the breakdown of ATP to ADP or AMP. The ratio of ATP to ADP of cells is a relative measure of energy stored in the battery, so this ratio must be maintained within relatively narrow limits despite rapid and sometimes large changes in the demands placed on metabolism. If something interferes with ATP production or accelerates ATP consumption to cause a decrease in the ATP–ADP ratio, mechanisms must exist for reestablishing energy balance and recharging the battery.
The model thought capable of explaining metformin’s action until very recently was based on activation of a critical sensor of cellular energy status that goes by the acronym AMPK (for AMP-activated protein kinase). When the concentration of AMP increases through the breakdown of ATP, AMPK switches on catabolic processes—such as the uptake and oxidation of glucose—that yield ATP, while at the same time switching off nonessential processes that consume ATP and downregulating several of the enzymes involved in the synthesis of glucose from simpler precursors.

Overall, metformin keeps blood glucose levels in check. For all intents and purposes, metformin diminishes insulin resistance by eliciting the accumulation of AMP, which activates AMPK and inhibits adenylate cyclase to accelerate glucose uptake from the bloodstream and its consumption in glycolysis, while at the same time putting the brakes on its production by gluconeogenesis. In short, metformin antagonizes the action of glucagon to cause a decrease in fasting blood glucose levels. Collateral effects of metformin include increased fatty acid oxidation and decreased glycogen, fatty acid, and cholesterol synthesis.
Illustration by Barbara Aulicino.
Elegant as it is in its explanatory power, it now appears that this model is unlikely to be the complete mechanism for one simple reason: Mutant mice that lack an active AMPK pathway do just fine and are as responsive as normal mice to metformin and other biguanides. The implication here is that instead of or in addition to acting through the AMPK pathway, metformin exerts its effects through another signaling pathway to antagonize the effects of the hormone glucagon. In doing so, metformin triggers a switch from glycolysis to gluconeogenesis, to make up for the glucose shortfall that is otherwise associated with starvation. If this is what actually happens, the bottom line is that in the final analysis metformin would have the same effect as insulin, in that it would accelerate glycolysis while at the same time decelerating gluconeogenesis. In other words, metformin would keep liver glucose output in check and, to all intents and purposes, correct the primary defect associated with type 2 diabetes by enhancing insulin sensitivity.
A final twist to metformin’s tangled story lies in the expansion of the drug’s applications to prediabetes and polycystic ovary syndrome (PCOS). If there are two conditions for which the benefits offered by metformin have yet to be fully tapped, it is probably these.
Not only will most prediabetics eventually progress to full-blown type 2 diabetes, but as is now also apparent, they will likely have already started to undergo cardiovascular deterioration. The stark truth of this condition, which affects some 30 percent of the U.S. population, is that many “normoglycemic” individuals have already lost half of their beta-cell function by the time they are deemed prediabetic and will have lost more than 80 percent by the time they hit the glycemic cutoff for type 2 diabetes.
The interest in metformin for the prevention or delay of type 2 diabetes in at-risk prediabetic subjects results from the ongoing U.S. Diabetes Prevention Program, which shows that overweight or obese subjects at high risk for type 2 diabetes can on occasion delay development of the disease for 10-15 years when prescribed metformin. That’s the good news. The not-so-good news is that only about 3.7 percent of patients with prediabetes are prescribed this drug because clinicians have no other choice than to prescribe it off-label pending FDA approval of it for this purpose.
Not only does metformin enhance insulin sensitivity, but it also lowers blood glucose and puts the brakes on weight gain and cardiovascular complications.
Another potential growth area for metformin, PCOS ranks as the most common endocrine disorder affecting women. Some 4-12 percent of women of reproductive age have this condition. The principal problem underlying PCOS is a hormonal imbalance associated with ovulatory and menstrual irregularity as well as androgen excess. The polycystic ovary component of the disorder arises from follicular progression without ovulation and from the formation of cysts. For some time, it has been appreciated that besides its reproductive repercussions, PCOS has a long-term impact on insulin resistance and its progression to type 2 diabetes and cardiovascular complications. Given that insulin resistance is considered to play a significant role in PCOS either directly or through obesity, which has an estimated prevalence among PCOS women of 60-70 percent, the need to address this facet of the condition through the deployment of agents such as metformin is now a prime clinical concern.
Researchers in Venezuela first explored metformin therapy as a treatment option for PCOS in 1994. This study reported a significant improvement in the regularity of the menstrual cycle and a reduction in circulating insulin and androgen levels, together with a marked decrease in insulin resistance and body weight. Now in both the United Kingdom and United States, women diagnosed with PCOS who are insulin resistant are advised to take metformin to encourage fertility and control the other symptoms of the disorder. But because metformin is not licensed for treating PCOS by the National Institute for Health and Clinical Excellence (NICE) in the UK nor approved for this condition by the FDA in the United States, it must be prescribed off-label in both countries.
Even this expansion of the use of metformin for PCOS as well as prediabetes and diabetes is quite possibly not the end of the road. A particularly noteworthy irony of the metformin story is that the drug whose rite of passage was to prove so tortuous may have collateral benefits—which likely would not have been realized had it not been prescribed to so many—above and beyond those for which it was eventually to get a footing.
There are now indications that the merits of metformin may extend to cardioprotection and cancer prevention. Long-term treatment with metformin seems to confer a degree of cardiovascular protection, supplemental to the benefits associated with better blood glucose management, so as to diminish the likelihood of myocardial infarction, heart failure, and diabetic cardiomyopathy. Although the precise mechanisms underlying these effects are unknown, several have been proposed, including metformin-elicited decreases in oxidative stress in combination with diminished insulin resistance in the cells lining the inner surfaces of blood vessel walls, as well as those from which heart tissue is built.
On the cancer front, the results of epidemiological studies first published in 2005 point to the possibility that diabetics treated with metformin have a lower incidence of cancer than diabetics treated with other agents. Similar conclusions have come from several subsequent studies. The mechanistic basis of these effects and whether they are equally applicable to nondiabetics is currently an area of considerable research activity (and controversy), with an eye to the deployment of metformin and other biguanides as cancer prophylactics and perhaps even as drugs repurposed for the treatment of cancer.
In recounting the fitful twists and turns of metformin’s discovery and implementation, we are repeatedly reminded of what the French philosopher and humanist Voltaire (1694-1778) penned more than 250 years ago: “Doctors are men who prescribe medicines of which they know little, to cure diseases of which they know less, in human beings of whom they know nothing.”
Click "American Scientist" to access home page
American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.