How Bacterial Pathogens Emerge
By Salvador Almagro-Moreno
Can scientists predict where disease-causing microbes will arise before they cause the next pandemic?
Can scientists predict where disease-causing microbes will arise before they cause the next pandemic?

In the blink of an eye, our world can be turned upside down by the appearance of a new pathogen such as SARS-CoV-2, the virus that causes COVID-19.
These devastating events are not something new to humanity, and, unfortunately for those of us trying to forecast them, they do not typically occur in a slow and predictable fashion: Harmless organisms can undergo quantum leaps in evolution to become deadly and then spread like wildfire. The plague and cholera have killed millions over the past centuries and are ominous examples of this phenomenon. Both diseases are caused by bacteria that began with an innocuous ancestor but evolved to become some of humanity’s worst scourges.
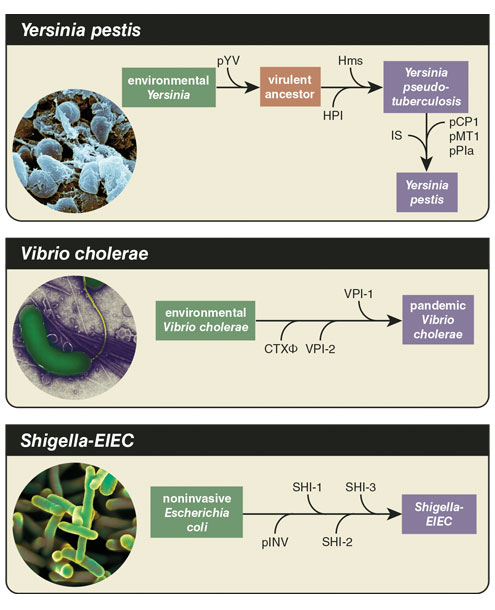
We cannot predict the future, but we can learn many critical lessons from studying the past of pathogenic disease. Black death reached pandemic status after Yersinia pestis, the bacterium that causes bubonic plague, experienced two major sequential changes. Y. pestis emerged from its ancestor, Yersinia pseudotuberculosis, about 5,000 years ago somewhere in the Eurasian continent, leading to a drastic change in its disease profile from a relatively benign intestinal pathogen to a deadly systemic one capable of causing pandemics. After Y. pestis acquired one cluster of genes, it gained the ability to cause the respiratory form of the disease. Another leap, the acquisition of a gene called ymt, gave rise to transmission via fleas.
This seemingly minor change gave the bacteria the ability to cause bubonic plague and spread both within a human body and among humans in a way that changed human history: The Black Death devastated the Eurasian continent from 1347 to 1351, leading to Europe losing over a third of its population. Vibrio cholerae, the agent of the severe diarrheal disease cholera, experienced a similar transition from a harmless ancestor to one of humanity’s worst curses. The evolutionary history of this aquatic bacterium involves a few more players than Y. pestis, most of them clusters of genes acquired from the environment.

Science History Images/Alamy Stock Photo
The events that can lead microorganisms such as these to turn from benign to bad are as diverse and numerous as the pathogens themselves. Over the past several decades, scientists have uncovered myriad mechanisms and factors that mediate this critical transition. These findings are helping us develop more sophisticated frameworks to address emerging pathogens before they become serious problems for public health.
The process by which a biological agent that poses no threat to human well-being gains the ability to colonize and harm us is called pathogen emergence. Typically, this process happens when a microorganism with an environmental lifestyle that does not need people to exist, or a commensal one that harmlessly grows on or within us, becomes capable of harming us.
The emergence of novel human pathogens is without a doubt one of the most pressing problems that we face. I have been working for almost two decades trying to understand what the biological rules and evolutionary forces are that make a microorganism transition to causing disease in humans. It all started when I was a graduate student at the National University of Ireland working to identify large pieces of DNA called pathogenicity islands that differentiate pathogenic V. cholerae from its more innocuous cousins. Between genomic comparisons, DNA extractions, and pints of Murphy’s, my curiosity to understand pathogen emergence grew, and I have dedicated my career ever since to investigating this complex phenomenon.
When I started studying pathogen emergence, the field was still relatively in its infancy. The leading view at the time was that harmless organisms acquired discrete pieces of DNA that conferred univocal properties such as the production of a toxin or the expression of a critical factor that allowed bacteria to colonize the human host. Over the years, novel findings have provided a larger context for these entities, unearthing previously unrecognized relationships between these elements and the bacterial genome. Furthermore, my group and others are identifying the ecological drivers that lead to the selection of pathogenic traits and virulent strains. The connections exposed by these discoveries are beginning to provide the tools we need to potentially forecast the events and drivers that lead to the emergence of novel infectious agents, which is pivotal for successful disease management and control.
Pathogen emergence is a dynamic, complex, and multifactorial phenomenon that involves an interplay of various genetic adaptations and ecological drivers. Microorganisms are built to evolve. Bacteria in particular, such as the agents of cholera and the plague, possess incredibly sophisticated mechanisms that render them capable of acquiring pieces of DNA from the environment or accumulating mutations that allow them to adapt to new environments in a way that multicellular organisms such as humans never could. They are essentially “evolution machines.”
Stephanie Freese
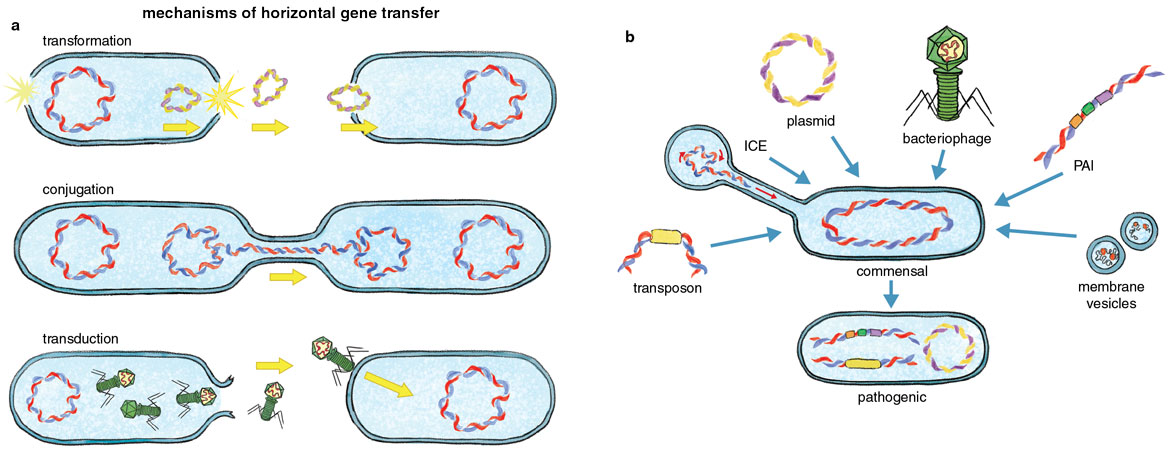
One process that has vastly influenced pathogen emergence is the acquisition of new genes via horizontal gene transfer. Humans can only pass genes vertically through generations, that is, from parents to offspring. In contrast, bacteria can pass on genes vertically and also horizontally, trading chunks of genetic information among one another in ways that can accelerate evolution. Through this type of gene transfer, some virulence traits emerge when a cell acquires genetic material from other microorganisms or from its surrounding environment. These traits are often encoded in mobile genetic elements, such as plasmids, pathogenicity islands, or bacteriophages, which are horizontally acquired factors that move around in the environment and play a critical role in the emergence of pathogens. Understanding the complexity and wealth of these mechanisms will help reveal the potential ways we can detect emerging pathogens and develop surveillance platforms.
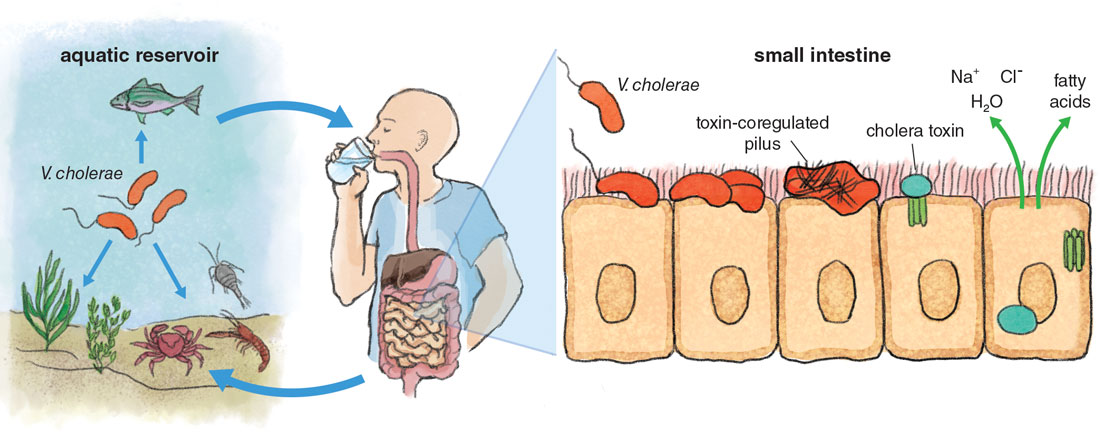
For instance, plasmids are circular pieces of DNA that can be acquired from the environment via a process of gene uptake called transformation and have long been associated with the attainment of pathogenic traits such as toxins and antibiotic resistance. Plasmid acquisition has even led to events where new species of pathogens emerge, including the emergence and speciation of the pathogenic Yersinia species from environmental strains. Acquisition of the virulence plasmid pYV by environmental Yersinia results in a virulent ancestral strain that can overcome host defense mechanisms and colonize lymph tissues. Pathogenicity islands are large clusters of genes that are confined to pathogenic strains within a species and typically encode important genes that are critical for host colonization. For instance, the pandemic strains of V. cholerae harbor several major pathogenicity islands; one encodes an essential intestinal colonization factor called toxin-coregulated pilus. Bacteriophages are viruses that can infect bacteria and in certain cases can integrate within the genome and confer new traits to the host cell. Bacteriophages are a major vehicle for the acquisition of bacterial toxins in several pathogens. Bacteriophages led to the emergence of new virulent and epidemic clones of Escherichia coli 0157:H7, a foodborne pathogen that causes hemorrhagic diarrhea, and these viruses also conferred the cholera toxin to pathogenic strains of V. cholerae, the source of the diarrhea associated with cholera.
Not all of these elements are acquired directly from the surrounding environment. For instance, integrative and conjugative elements are transferred via conjugation, a process that requires the production of an extracellular appendage (called a conjugative pilus) that connects two bacterial cells and allows them to exchange genetic material. Integrative and conjugative elements typically encode genes that provide resistance to antibiotics, help outcompete other bacteria during colonization, and enhance survival of the bacterium within the host. Insertion sequences and transposons are small “jumping” elements that can “jump” within the genome and insert in different locations. Transposons primarily move around genes associated with antibiotic resistance. Interestingly, unlike pathogenicity islands or plasmids that physically carry and transfer virulence-associated genes, insertion sequence elements primarily disrupt genes by inserting in them. This literal movement of DNA leads to genome rearrangements that alter gene expression. Integrons are genetic elements encoded within the ancestral bacterial chromosome that facilitate the efficient capture and expression of genes that have been acquired from the environment. Integrons primarily capture genes associated with antibiotic resistance, but some of the genes they contain can also drive host colonization and virulence.
Stephanie Freese
In addition to these canonical modes of horizontal gene transfer, other gene transfer mechanisms have been identified that might play significant roles in the emergence of bacterial pathogens. Membrane vesicles are extrusions of the cell membrane resembling little spheres that get secreted into the environment, carrying cell content and, importantly, DNA. It was recently found that membrane vesicles from pathogenic E. coli can harbor genes encoding virulence factors that result in increased cytotoxicity of nonpathogenic E. coli strains. Recently discovered gene transfer agents also encompass another potential mode of horizontal gene transfer. These elements exclusively package random segments of bacterial DNA and inject them into recipient cells. Future research will uncover the impact of these novel transfer mechanisms in shaping the emergence of bacterial pathogens and potentially reveal novel emergence mechanisms, which will undoubtedly enrich and likely reshape our picture of what it takes to become a pathogen.
Clearly, a plethora of mechanisms and vehicles exist that allow bacteria to acquire new traits from the environment, helping them achieve the quantum leap that can lead to their emergence as pathogens. But evolution does not follow an intentional course, so the quantum leap can also take bacteria in the wrong direction: Acquiring foreign genetic fragments can cause substantial wreckage and disrupt an organism’s well-ingrained physiology. Identifying the environments and conditions that favor microbial risk-taking is an active area of research.
The emergence of novel human pathogens is without a doubt one of the biggest problems that we face.
It turns out that bacteria are very picky about the genes that they acquire and express. This genetic shuffling is held in check by a variety of intricate cellular processes. Pathogens must carefully balance the acquisition and expression of the genes that could potentially help them colonize the human host against the indiscriminate uptake and assimilation of detrimental genes that would compromise their survival. To do so, they have devised complex ways to modulate DNA acquisition and gene expression. In contrast to integrons, which favor the successful maintenance and expression of acquired genes, DNA assimilation is inhibited by either degrading incoming DNA via CRISPR-Cas systems, or by preventing gene expression via xenogeneic silencers (“xenogeneic” means obtained from an organism of a different species). CRISPR-Cas systems have gotten substantial media attention as a new technology to edit genomes; however, their original function is as a bacterial defense mechanism that can degrade incoming pieces of foreign DNA. (See “Interview with a Gene Editor,” July–August 2015.)
Interestingly, CRISPR-Cas systems have recently been found to prevent the establishment of specific gene clusters associated with pathogen emergence. For instance, during mice infections with Streptococcus pneumoniae, CRISPR-Cas sequences that target the capsule genes prevent this respiratory pathogen’s cells from becoming encapsulated, eventually blocking establishment of a successful infection. These findings could help explain why many strains of major human pathogens lack CRISPR-Cas systems in their genomes, as they naturally prevent the acquisition of foreign genes.
Gene silencing is another widespread approach for bacteria to control the expression of new genes potentially associated with virulence. For this mechanism, bacteria use xenogeneic silencing proteins that bind to and suppress the expression of these acquired genes. This mechanism minimizes the potentially negative effects associated with unregulated expression of foreign DNA. Proteins such as HN-S in E. coli are critical regulators of acquired virulence genes, and proteins with similar function and evolutionary origin can be found in a wide range of important human pathogens such as Mycobacterium tuberculosis or Pseudomonas aeruginosa.
If all it takes to emerge as a pathogen is to acquire some mobile genetic elements, why do only a small number of species and strains cause disease in humans? For instance, the mobile genetic elements that confer pathogenic traits to V. cholerae can be easily exchanged and are widespread in cholera endemic areas. In fact, numerous environmental nonpathogenic strains of V. cholerae encode these islands and the bacteriophages required to develop the disease but are totally harmless. However, only one group, the aptly named pandemic group, can cause cholera in humans. What is so special about them?
Barbara Aulicino; Cultura Creative Ltd/Alamy Stock Photo; Dennis Kunkel Microscopy/Science Source
This question puzzled me throughout graduate school, where I was investigating the role of horizontally acquired DNA in V. cholerae, but it wasn’t until my time as the Ernest Everett Just Postdoctoral Fellow at Dartmouth College that I started to explore it fully. The only way I could make sense of the data was under a scenario in which those strains that emerged as pathogenic had a unique genetic makeup that provided “fertile ground” for these mobile genetic elements to land on. A few years later when I started my own lab, we discovered that strains that cause cholera encode what we call “virulence-adaptive polymorphisms,” which are variations that provide them with preadaptations to virulence. These preadaptations are independent from the horizontal acquisition of genetic material and occur in the environment prior to the bacterium acquiring the ability to colonize the human host.
This behavior is not unique to pathogenic strains of V. cholerae. Other studies have explored the relationship between the genomic background of E. coli strains and the retention and expression of acquired virulence factors. These analyses have identified two distinct genomic profiles in pathogenic strains: first, an ancestral background that allows expression of factors associated with mild, chronic diarrhea and is found in most E. coli, and second, a derived background that favors expression of factors with more severe pathologies and that is found in pathogenic strains such as enterotoxigenic or enterohemorrhagic E. coli. It is becoming increasingly clear that the specific genomic makeup of a given strain is essential for the emergence of pathogenic traits and its ability to colonize the host, which might explain the uneven distribution of virulent clones in numerous pathogenic species.
Besides a particular set of preadaptations in the genomic background of pathogens, there are mutations that arise within the host that also confer virulence-adaptive traits, also known as pathoadaptive mutations. These mutations are typically associated with the colonization of different tissues, adaptation to new metabolic conditions, or evasion of the immune system. A quintessential example of the significant role of pathoadaptive mutations in pathogen emergence is the rise of pathogenic Shigella species, water and foodborne pathogens that cause severe diarrhea, from their closely related nonpathogenic commensal E. coli. The latter require the activity of an enzyme called CadA-mediated lysine decarboxylase for growth in the intestine; however, CadA blocks the activity of the Shigella toxins. Pathoadaptive mutations in the cadA gene occurred independently in various Shigella species, allowing for toxin activity and contributing to the emergence of pathogenic Shigella from commensal E. coli.
These mutations also allow pathogenic E. coli to colonize new tissues and adapt to new niches. For instance, more than 90 percent of commensal E. coli express a gene called mannose-sensitive type 1 fimbriae that helps them colonize the intestine. Pathoadaptive mutations in this gene help the bacteria adhere more tightly to the lining of the urinary tract, allowing a change in the type of tissue that E. coli can colonize. This change ultimately leads to a major evolutionary transition from an intestinal commensal to a pathogen that causes urinary tract infections. The emergence of other major virulence phenotypes such as resistance to host defense mechanisms is also associated with pathoadaptive mutations. For instance, commensal Mycobacterium abscessus strains that are adapted to the lungs of cystic fibrosis patients possess a large number of mutations in genes that lead to enhanced survival within macrophages.
All these examples highlight the relevance of unique genomic adaptations in the process of pathogen emergence. These, together with horizontally acquired DNA, appear to be main molecular drivers of this complex phenomenon. Much of my research has revolved around a key question: What evolutionary forces select for these disease-causing traits?
A major driver of the evolution of pathogenic bacteria is predation. In the wild, bacteria must survive assaults by bacteriophages, protozoa such as amoeba, and predatory bacteria such as Bdellovibrio. The evolution of a variety of defense mechanisms against these threats has been accompanied by the incidental development of virulence traits that aid in the colonization of humans. For example, some of the defense mechanisms in Legionella pneumophila, E. coli, and P. aeruginosa that are critical for evading predation by protists in their natural environment are also fundamental for the ability to colonize and survive within human hosts.
A plethora of mechanisms allow bacteria to achieve the quantum leap that can lead to their emergence as pathogens.
L. pneumophila, which causes the severe respiratory syndrome Legionnaires’ disease, represents a fascinating case of accidental evolution of virulence. Survival and spread of the bacterium in freshwater reservoirs and moist soil are contingent upon replication in free-living protozoa such as Acanthamoeba castellani. The pool of virulence traits in this pathogen evolved as a result of its interaction with aquatic protozoa, which in turn enabled its ability to survive within human alveolar macrophages. This relationship is so well established that the bacterium has acquired a large number of genes from its eukaryotic host. For instance, the enzyme in L. pneumophila sphingosine-1 phosphate lyase (LpSpi) plays a role in sphingolipid biosynthesis, a pathway that is conserved in protozoa but is hardly ever found in bacteria. LpSpi modulates the metabolism of the protozoa to aid L. pneumophila growth by preventing cell-death processes that naturally occur upon infection. Interestingly, the gene is related to their eukaryotic counterparts, suggesting horizontal gene transfer from A. castellani to L. pneumophila.
Legionella species are not the exception, but rather some of the many examples in the bacterial world of acquiring genes from eukaryotes. Amoebae may be training grounds for future bacterial pathogens as the ecologically and phylogenetically diverse bacteria thrive inside amoebae, acquiring traits that also allow for survival and pathogenesis in the human host. Therefore, it is evident that the environment and its dwellers can serve as selective agents that ultimately lead to the emergence of bacterial pathogens. As humans alter that environment, we may be easing their path toward colonizing us.
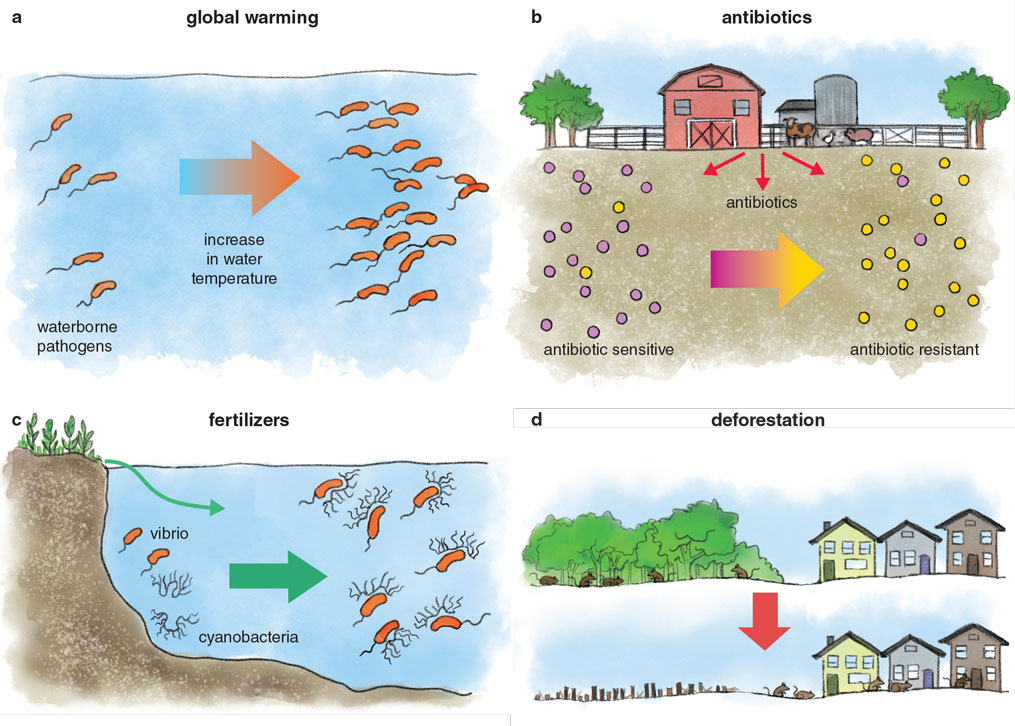
Human-made ecological perturbations, such as climate change or pollution of our ecosystems, are drastically affecting the spread and proliferation of disease-causing bacteria, accelerating their rampant acquisition of antibiotic resistance, and increasing the likelihood of the emergence of novel pathogens. The warming of the oceans is leading to a measurable increase in the overall number of Vibrio bacteria and their associated infections over the past few decades. Antibiotic resistance is drastically increasing among environmental bacteria, in large part because of the addition of antibiotics to farm feed and treating orchards with antibiotics to prevent blights. The runoff of fertilizers due to heavy rains leads to cyanobacterial blooms in estuaries and lakes. We recently found that those blooms lead to the proliferation and fast growth of vibrios. Interestingly, some studies have shown a direct correlation between cholera cases and the presence of these blooms in the Bay of Bengal.
Stephanie Freese
Land-use changes, such as deforestation, urbanization, and agricultural expansion, disrupt biodiversity and the food-web structure, and cause habitat fragmentation. The resulting loss of habitat introduces non-native species, including pathogens, into the human matrix, thus increasing host-pathogen interactions, ultimately leading to the emergence of new diseases in humans, domestic animals, and wildlife. For instance, deforestation and land disturbances in the southern Amazon basin of Peru have been correlated with the growing prevalence of rodent-borne Leptospira and Bartonella species. Forest fragmentation in the northeastern United States has led to an increased risk of Lyme disease due to an abundance of white-footed mice, considered “superspreaders” of the tick-borne pathogen Borrelia burgdorferi. Deforestation activities in Brazil have been shown to influence the risk of Brazilian spotted fever caused by the vector-borne bacterium Rickettsia rickettsii.
Spillover of zoonotic pathogens from animals to human hosts, such as the transition of SARS-CoV-2 from bats to humans, accounts for the majority of the emerging infectious disease events in the past 80 years, more than half of which were of bacterial origin. However, besides research on how environmental perturbations affect the spread or antimicrobial resistance of pathogens, there have been very few studies investigating how these changes affect the likelihood of a bacterium becoming pathogenic or drive the acquisition of novel pathogenic traits. This is a major gap in our current knowledge that, once addressed, will lead to unprecedented advances in our ability to predict and identify sources of potential pathogens.
Pathogen emergence is a dynamic and complex phenomenon, which makes it enormously challenging to study. For instance, which variables do we study to attempt to find associations? If we find one variable, how do we switch from correlation to causation? How often and how many samples do we need to generate accurate predictive models? Most importantly, is it possible to build a realistic and cost-effective surveillance framework?
Effective prediction of emergence events is now a possibility and will benefit disease management and public health measures.
My lab members and I recently aimed to answer some of these questions by implementing a holistic approach where we mixed ecology, computational biology, and molecular genetics to dissect the drivers that foster the selection of virulence traits and pathogenic clones within environmental populations. We focused on Vibrio vulnificus, an aquatic bacterium that can cause a deadly flesh-eating infection in humans, as a model system. Our findings, which were published in the Proceedings of the National Academy of Sciences of the U.S.A., suggest how ecosystems generate selective pressures that facilitate the emergence of specific strains with pathogenic potential in a natural population. These insights can be applied toward predictive frameworks to assess the risk of pathogen emergence from environmental sources. Effective prediction of emergence events is now a possibility and will benefit disease management and public health measures.
Courtesy of Salvador Almagro-Moreno
Sequencing technologies are becoming more affordable, and their widespread implementation is helping address the roots of pathogen emergence. For instance, techniques that allow us to examine the microbial communities in a given environment and their evolution can be used to determine the impact of natural and anthropogenic-induced effects on ecosystems. These approaches can also identify risk factors such as the increased prevalence of bacterial strains with pathogenic potential in a population, or the availability of known virulence factors in environmental reservoirs. The data obtained using these techniques can provide valuable insights into the likelihood of new emergence events. Furthermore, the integration of this genomic information paired with the currently used culture- or PCR-based monitoring of natural habitats will significantly strengthen surveillance platforms to detect pathogen emergence by providing an in-depth yet affordable means of monitoring potential pathogen sources. Such informed surveillance networks and subsequent risk factor analyses will help us identify and categorize high-risk habitats, such as prioritizing specific sites and seasons for sampling rivers and lakes, identifying certain animals and meat that are of concern in markets, and monitoring specific rodent or insect populations. As a result, these efforts can lead to appropriate interventions and public health advisories for effective outbreak prevention and control.
In addition to potentially helping forecast pathogen emergence events, these approaches can also be useful after an emergence event or outbreak. For instance, retroactive outbreak analyses on the contributing environmental and genetic factors can be informative in preventing future emergence events. We recently applied this approach to the catastrophic cholera epidemic that killed thousands of people in Latin America in the 1990s, gleaning important lessons, including the role of weather patterns such as El Niño and the importance of specific food preparations, which can be applied to future outbreaks. Despite the numerous dark events in human history caused by emerging pathogens, we and other scientists are gathering the knowledge, tools, and approaches we need to help us detect the next quantum leap in pathogenicity, and potentially avert the next pandemic.
Click "American Scientist" to access home page
American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.