Alzheimer's Disease: The Great Morbidity of the 21st Century
By Charles T. Ambrose
Neuroangiogenesis (NAG) provides a vascular basis for understanding Alzheimer’s disease, senile dementias and cognitive decline with aging.
Neuroangiogenesis (NAG) provides a vascular basis for understanding Alzheimer’s disease, senile dementias and cognitive decline with aging.

DOI: 10.1511/2013.102.194
Alzheimer’s disease bids to become in its own way as devastating in the foreseeable future as any of the great plagues of the past. The Black Death (bubonic plague of the 14th century) was also called the Great Mortality because it killed many of those infected within a week or two of its onset. By analogy but in contrast, Alzheimer’s disease could be termed the Great Morbidity, because it sends affected persons down a fading mental spiral for decades before their death from other causes.
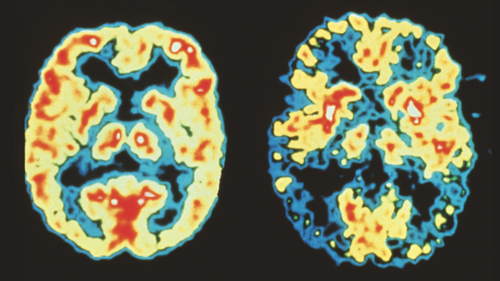
Dr. Robert Friedland/Science Photo
In the United States at present, Alzheimer’s disease (AD) afflicts 13 percent of people age 65 and older and 43 percent of those 85 and older, or in total, an estimated 5.4 million—equivalent to the current population of Minnesota. AD is the fourth leading cause of death in the United States today. By 2030 the number of persons in the country with AD is expected to reach 7.7 million and by 2050 will approach 11 to 16 million—think of Ohio, Illinois or Pennsylvania. The impending economic and social impact from AD will compromise American life immensely during the coming decades … “unless preventative strategies are found,” as urged by numerous Alzheimer researchers.
Relevant in this discussion is the condition senile dementia (SD), which some regard as distinct from AD but others employ as a generic term encompassing it also. Throughout the greater world, senile dementia affects 5 to 7 percent of people over age 60. This number is expected to double in the next 20 years and reach 100 million by 2050.
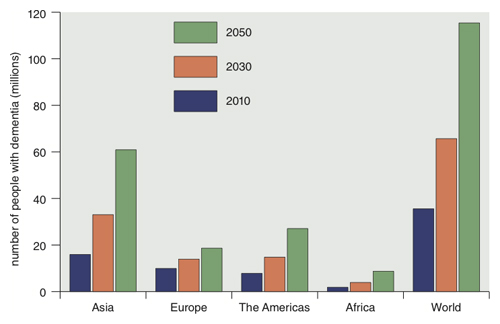
Data from Dementia: A Public Health Priority, World Health Organization 2012
In the literature are several other terms for progressively impaired mental states—for example, adult onset dementia, cognitive decline with aging, vascular dementia, and frontal temporal dementia. Each has a particular meaning, which need not be explained here. AD accounts for 50 to 70 percent of all dementias. I link all the above mental conditions together because they may have a common basis—that is, diminished cerebral capillaries. This essay is a shortened version of a longer paper in the Journal of Alzheimer’s Disease, 2012, which presents in more detail the NAG hypothesis to explain these conditions.
In 1906 Alois Alzheimer (1864–1915) reported a single case study of a female with presenile dementia, whose brains showed a massive loss of neurons, gummy deposits and strongly staining fibrils. He later found the same histopathology in the brains of three other senile patients. These cases were first described in a 1910 text book on psychiatry, where the condition was given the eponym Alzheimer’s disease. But this name appeared only sparsely in the indexes of books on internal medicine before the 1950. Nor did these same texts include much about senile dementia, other than in association with neurosyphilis or schizophrenia. As a relevant aside, my father’s medical school text (Cecil and Kennedy, 1929) contained only two short sentences about Alzheimer’s disease (22 words), whereas my text (Cecil & Loeb, 1951) omitted AD entirely. The question arises whether before 1950 senile dementias (including AD) were so uncommon that they warranted little mention.
Gregory Zilboorg, a medical historian, reviewed the very old medical literature for reports of senile psychoses/dementia. Aretaeus of Cappadocia, a 2nd-century C.E. Greek in Rome, remarked that “mental decay is a calamity of old age—without intermittence and incurable.” Rhazes, a 9th–10th century Persian, viewed melancholy as inevitable “in the lives of old and decrepit persons.” Felix Platter, a 16th-century Swiss physician, observed that the memory of some elderly people is defective, especially for recent happenings. And Jean E. D. Esquirol (1772–1840) described the mental changes occurring with old age—a waning memory (especially of recent events), a shorter attention span, progressive fatigue, slower movements, irritability and unreasonableness.
If mental deterioration in the aged has been noted in the past but paid little attention in early 20th-century medical texts, what accounts for the recent clinical prominence of AD and its predicted great increase? Some suggest it is better recognition of late. But another explanation offered is that the current U.S. population is older than previous ones and thus has had more years for geriatric conditions to appear before death. This latter notion does not seem well supported by data comparing the Alzheimer’s death rate per 100,000 people (AD-DR) in countries with different life expectancies (LE) but comparable levels of diagnostic expertise. The United States has an LE of 75.6 years and an AD-DR of 24.6, whereas in Switzerland the LE is 79.3, but the AD-DR is only 20.0. Similarly, Finland has an LE of 75.9 and AD-DR of 34.9, whereas in nearby Sweden people live three years longer (78.9), but the AD-DR is only 21.9. Genetic differences among populations may dampen the effect of aging. In any case, research into other explanations seems warranted.
Indeed, investigators have searched for environmental factors to explain the apparent increased incidence of AD after World War II—aluminum in cooking vessels, atmospheric pollutants and so forth. But none has proved to be a convincing inciting cause. Based on the apparent recent rising incidence of AD, a putative contributing factor may have appeared around the mid-1950s and prevailed since. Radio executives and some educators might like to incriminate the stultifying effect of TV, while classic music lovers might blame the brain-deadening effect of raucous rock music, but I’m not persuaded. Recently epidemiologists have invoked “risk factors” (biomarkers) contributing to the development of AD—for example, obesity, diabetes, hypertension, head injuries, lower intelligence, less formal education, inactivity (both physical and mental), certain alleles and so on. The search for the etiopathology of AD over the past several decades has produced an enormous number of studies, of which only a very small fraction has been included in the following brief review.
Alzheimer’s disease has been diagnosed on two levels—histologically by the presence of amyloid plaques (AP) and neurofibrillary tangles (NFT) in the brain and/or, in their absence, clinically by key cognitive symptoms and signs—loss of memory, orientation and reasoning. The pathogenesis of AD is likely multifactorial, entailing several or many sequential cellular and biological interactions. The ultimate defect is impaired synaptic connections due to neurons altered in physiology or reduced in number or size. Many investigators of AD have focused their attention instead on the penultimate cause—that is, on various physical or biochemical changes in the brain – for example, the presence of AP and NFT.
For several decades the amyloid cascade theory proposed by Hardy and Higgins in 1992 has dominated research into the etiopathology of AD. The production and assembly of these aberrant protein deposits have been extensively explored and postulated to act as foreign bodies in the brain, inducing there an inflammatory response with neuronal damage and death. AP first appears in the brain as an amyloid precursor protein (APP), which is cleaved by an APP secretase into the amyloid beta protein (Aβ). AP has been postulated to have a toxic effect on neurons directly or, when aggregated, via free radical formation, which leads to “oxidative stress” and the resulting degeneration and death of neurons. NFT consists of a hyperphosphorylated protein (tau), which destabilizes the neuronal cytoskeleton and aggregates into clumps/tangles. Numerous studies have concerned the clearance of AP across the blood brain barrier and Aβ/APP lowering drugs, whereas others have identified genetic alleles controlling AP. It’s not clear whether the presence of AP or NFT is a primary, penultimate event or secondary to some underlying process.
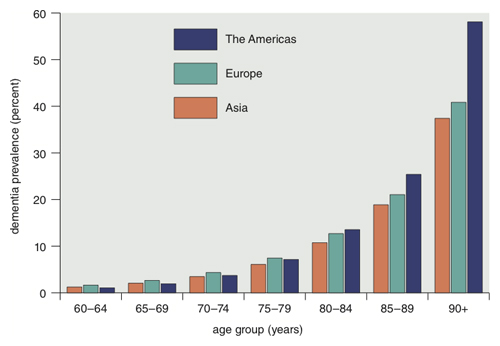
Data from Dementia: A Public Health Priority, World Health Organization 2012
Two troubling reservations about the amyloid theory are that some persons without symptoms of AD have many cortical Aβ deposits and that, as John Hardy and Dennis Selkoe put it, “the number of amyloid deposits in the brain does not correlate well with the degree of cognitive impairment” experienced by patients. This suggests that some other primary factor or condition is responsible for AD but may promote AP and NFT formation secondarily.
Other investigations have centered on the deficiency of acetylcholine in AD brains or on the excess of glutamic acid in nerve cells with a resulting calcium influx. Elemental aluminum, iron and mercury in the brain have been postulated as catalysts for free radical generation and the oxidative stress mentioned above. Protein oxidation, lipid peroxidation and other biochemical aberrations have been found in AD brains, as well as changes in the capillary basement membrane and glia. Scores of genes have been identified that influence early onset versus late onset forms of the disease. The literature is immense. But none of these areas of study of AD has provided a general unifying theory of its etiopathology.
An overview of AD might focus on three levels—genetic differences (which ultimately determine most aspects of brain biology/pathology), the proximate cause (toxic agents, vascular defects and so on) and peripheral influences (for example, risk factors). This essay concerns mainly one particular proximate cause—a diminished level of neuroangiogenic factors, as explained next.
The neuroangiogenic (NAG) hypothesis proposes that Alzheimer’s disease and the various dementias of the aged have in common a reduced microcirculation in key regions of the brain due to a waning number of cerebral capillaries with aging. The level of neuroangiogenic factors there is less than that needed to maintain a full complement of functional capillaries.
My interest in impaired cognition with aging stemmed from an earlier fascination with the localized motor skills highly developed in intensely trained musicians. In a 2010 article in American Scientist I proposed that the remarkable finger/hand/foot dexterity displayed by concert pianists is due not only to augmented synaptic connections in their primary motor cortex but also to an increased local capillary density (CD)—initiated and maintained there by continuing angiogenesis. Considerable data from the nonmusical neurological literature support this idea. I reasoned that if an increased local CD could account for remarkable motor skills of pianists, a decreased CD elsewhere in the brain might cause impairment—for example, the declining cognition of many aged persons.
This essay continues in two sections: The first concerns cerebral capillary density (CCD), whereas the second treats the formation and maintenance of these capillaries by neuroangiogenesis (NAG). In both sections the brain is analyzed during its three periods of life (during postnatal development, maturity and old age) and in cases of senile dementias (mainly AD). In each section, animal findings are cited before comparable ones in people. Next are summarized studies on brain-injured animals treated with angiogenic factors. The essay concludes with six approaches for administering such factors to persons with senile dementias.
Research on cerebral microvascularity began in the early 1920s with Edward Horne Craigie (1894–1989), a biologist at the University of Toronto, who examined male albino rats from their birth to 13 months of age (day 364). He measured cerebral capillary density in various parts of their brains by counting certain structural features of the capillaries in thinly cut slices (2–6 micrometers thick), where obviously only segments of vessels remain. He used the sum of the lengths of fragments visible in a fixed area of each brain slice over a fixed number of slices, to yield a composite value, Lv in micrometers per cubic micrometer. To exclude arterioles, only vessel fragments with diameters generally smaller than 12 micrometers were included in his measurements. Later investigators employed other indices of capillary morphometry, as outlined in my Journal of Alzheimer’s Disease paper. Below are summarized CCD findings for the four periods outlined previously.

Photograph at left courtesy of the University of Toronto Archives. Photograph at right Mike Stewart/Sygma/Corbis.
Postnatal period. In young rats Craigie examined the five laminae (Brodmann’s layers) in each of five cortical areas for Lv and found that vascularity in all areas rose rapidly from birth (average of 306) to day 21 (average of 862) but only slowly thereafter.
In the human brain the most rapid growth occurs between ages 3 to 6 years in the frontal lobes and from 6 to 15 years in the temporo-parietal region. Diemer compared the capillary density in the frontal cortex of newborn infants and adults and found the former to be 98.4 capillaries per square millimeter and the latter to be 290 per square millimeter.
Maturity. After the brain’s full development, the functional status of the cerebral microcirculation depends on the maintenance and replacement of capillary endothelial cells. The turnover rates for these cells in animal or human brains have not been reported, but such values for endothelial cells elsewhere in the bodies of various animals are available from studies which employed thymidine labeling of capillary endothelium and autoradiographs of tissue sections. For example, in 1967 Engerman and co-workers found that 0.01 percent of the capillary endothelial cells in the adult mouse retina were labeled after a single injection of tritiated thymidine and interpreted this as a turnover time of three or more years. More recently, based on the “mean of 14 studies” mainly in mice by numerous investigators, Denekamp concluded that the “potential turnover time” of capillaries of “normal tissues” is 60 days. Accepting Denekamp’s mean value of six times per year as a possible turnover number in the human brain and assuming an average life span of 60 years after puberty, I calculate that cells in the cerebral capillary bed undergo replacement 360 times during the average adult’s life. On the other hand, if one adopts Engerman’s three-year estimate of capillary endothelial turnover, then these cerebral capillaries would undergo regeneration 20 times during an average adult lifetime. Whatever the true value, replenishment occurs and is mediated by angiogenic factors in the brain.
Senescence in rats. Craigie found that average value of cerebral capillary density in aged rats rose to a maximum by month 13 in three cortical regions but declined in the two others after month 3 or 5. In regio temporalis CCD rose to 958 micrometers in month 3 but was 880 micrometers by month 13. And in the regio insularis CCD peaked at 856 micrometers during month 5 but fell to 638 micrometers by month 13. Buchweitz-Milton and Weiss recorded values for Lv (sum of capillary lengths in millimeters per cubic millimeter) in the cerebral cortex of “young” rats (8–10 months) as 830 and of senescent rats (28–33 months) as 577. A similar decline in Lv was reported in all other brain areas of the older rats. Klein and Michel measured various neocortical components (neurons, glial cells, vascular tissue) in the frontal and occipital neocortex of young adult rats (6–8 months old) and aged ones (25–27 months old) and found that combined component counts were 448 in the young adult group and 314 for the old rats.
Senescence in people devoid of recognized dementia. Impairment of cerebral capillaries in older persons was observed by Stewart and colleagues. They obtained biopsy specimens from the neocortex and underlying white matter in patients ranging from 20 to 80 years in age who had undergone surgery for glial tumors or corticectomy for intractable epilepsy. As measured by electron microscopy, the wall thickness of capillaries in both gray and white matter was thinner in older subjects than younger ones. The authors interpreted this age-related thinning as due to “a net loss of endothelial cells, with resultant elongation of the remaining cells.”
A deceased cerebral capillary density was reported by William B. Abernethy and colleagues in the paraventricular nuclei of 19 older human subjects, who ranged in age from 30 to 85 years and had no history of psychiatric treatment. Somewhat related is an earlier study by Kuwabara and Cogan, who had noted a decline in the number of endothelial cells “in the capillaries of the peripheral retina in persons past middle life (50 years).”
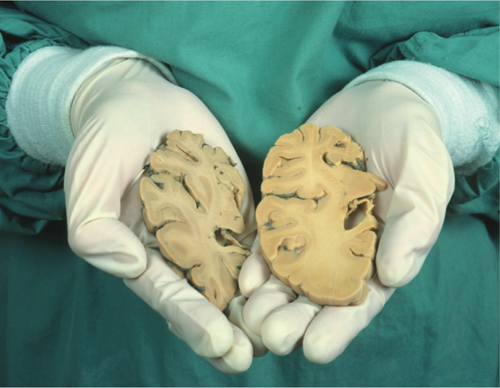
Senile dementias, mainly Alzheimer’s disease. Bell and Ball measured microvascular densities of capillaries and arterioles in the hippocampus of three groups: normal young persons (mean age of 38 years), normal old (74 years) and Alzheimer’s patients (78 years) and recorded the overall mean values as 129, 108 and 107 millimeters per cubic millimeter, respectively. Fischer et al. examined four areas in normal versus AD brains and found average vascular density indices (rounded off) as follows: basal forebrain, 87 versus 43; hippocampus, 82 versus 50; pre-frontal, 95 versus 75; and motor/sensory, 94 versus 83. And finally, Buée et al. reported the “percentage of the ratio capillary surface area/cortical … field area” for three groups: young controls, 26.32 percent; elderly controls, 18.95 percent; and AD patients, 16.50 percent. This subject has also been extensively reviewed elsewhere.
Isaac Chesar Michaelson (1903–1983), a British ophthalmologist, studied capillary growth in the developing retina of cats and humans, and in 1948 was the first to postulate the existence of a vasculogenic factor. In 1971 Judah Folkman (1933–2008) at Harvard Medical School isolated a tumor angiogenesis factor (TAF) from solid tumors whose growth depends “on the continuous build-up of a new capillary network.” Angiogenesis was soon recognized also to be a physiological response in various non-tumor conditions—for example, during embryonic and fetal development, in the formation of ovarian follicles and transiently in the corpus luteum during ovulation and during the development of the placenta. Neovascularization is a vital part of wound healing and fracture repair but ceases when these tissues are fully recovered. In contrast, uncontrolled angiogenesis promotes progressive solid tumor growth, occurs also during chronic inflammation and is associated with more than 70 human disorders, which Folkman grouped together as “angiogenic diseases”—for example, diabetic retinopathy, rheumatoid arthritis and psoriasis. Finally, angiogenesis determines CCD—its early development, maintenance and senescence, as discussed below.
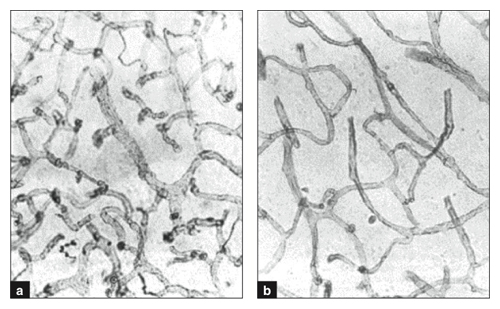
This image first appeared in 1990 and was reprinted in Vagnucci and Li in The Lancet 2003.
Folkman’s isolation of TAF in 1971 was followed by the discovery of other endothelial growth factors. Those listed below have been found in the brain. The first identified from non-tumor sources was the fibroblast growth factor (FGF). The one most abundant in the brain is vascular endothelial growth factor (VEGF)—originally called vascular permeability factor (VPF). Other brain-localized factors include epidermal growth factor (EGF), transforming growth factor-beta (TGF-β), platelet-derived growth factor B secreted by brain endothelial cells (PDGF-β) and angiopoietins (endothelial cell-specific cytokines). The various factors/cytokines associated with angiogenesis have been thoroughly reviewed. They are discussed below for the brain during its three ages and in the senile state.
Postnatal period. Breier et al. determined that vascular endothelial growth factor transcript levels (VEGF mRNA) in mice are “abundant in the ventricular neuroectoderm of embryonic and postnatal (six-day-old) brain when endothelial cells proliferate rapidly but [are] reduced in the adult when [as the authors state] endothelial cells proliferation has ceased.” I could find no studies on angiogenesis during human infancy.
Maturity. Two groups have used northern blot analysis to measure transcript mRNA levels during maturity. Breier and colleagues reported that VEGF mRNA levels in the choroid plexus of adult mouse brains “appeared slightly reduced” compared with that in six-day-old post-natal brains. Maxwell and ciolleagues examined the brains of patients with astrocytomas. In the adjacent normal brain tissue they found “a weak expression of TGF-α mRNA (transforming growth factor) and near background levels of EGF-R mRNAs (epidermal growth factor).
SPL/Science Source
The idea of a sustaining level of angiogenesis has been considered by others. Based on his studies in rats, Bär wrote that the maintenance of capillary plexuses or their repair “needs a continuous action of an angiogenetic stimulus.” In discussing angiogenesis inhibitors in human beings, Carmeliet concluded that there are “threshold levels of VEGF for the survival and maintenance of quiescent vessels in healthy organs.”
Senescence. Black and co-workers measured the capillary density in the visual cortex of rats housed under different environmental conditions (standard versus enriched) and concluded that there is “a failure of angiogenesis in unstimulated and old rats.” While angiogenic growth factors have been identified in adult/mature human brains (see above), I found no data on levels in neurologically healthy aged persons.
Senile dementias, mainly AD. Below are four reports on the level of endothelial growth factors or their mRNAs in Alzheimer’s patients.
In 1998 Kalaria and colleagues examined the brains of 12 AD subjects and found prominent VEGF reactivity in 40 percent of the cells resembling astrocytes. The group reasoned that this increased VEGF reactivity, which was “often localized around vessels,” might “compensate for insufficient vascularity and reduced cerebral perfusion in AD.”
In 2004 Yang et al. wrote that “VEGF is co-localized with Aβ plaques in the brains of patients with AD … most likely results in deficiency of available VEGF … [and] may contribute to neurodegeneration and vascular dysfunction in the progression of AD.”
Growth factors other than VEGF have been studied by Stopa and team in demented and aged human brains. Increased levels of basic fibroblast growth factor were found in association with the neuritic plaques and neurofibrillary tangles in AD brains. The sequestered bFGF may represent a decreased “bioavailability to surrounding cells.”
Similar studies by Styren et al. showed increased levels of epidermal growth factor receptor on brain vascular endothelial cells of aged and demented persons (many with AD) as compared with neurologically normal subjects. EGFR was interpreted here as a “proliferative signal in response to cellular injury … damage to the nervous system or to the brain vasculature itself.”
Folkman wondered whether purified angiogenic factors could “be administered in vivo, either locally or systemically, to accelerate the healing of wounds and fractures, or to increase neovascularization in the ischemic or infarcted heart.” The following four reports (from among others) describe growth factors being administered onto or into the brains of brain-injured animals.
Three laboratory groups subjected rats to middle cerebral artery (MCA) occlusion by different methods and introduced recombinant VEGF165 by various routes—onto the brain surface or into lateral ventricles. The infarct volume was measured in all brain sections and found to be greatly reduced in the treated animals compared with control animals. Hayashi suggested a protective action on the capillaries of the factor absorbed from the Gelfoam applied to the brain surface. Zang postulated the formation of newly grown capillaries. Sun concluded that VEGF reduced the infarct size in his rats and enhanced cerebral angiogenesis and neurogenesis. Similar studies by Kanya and by Wang in mice involved MCA occlusion and treatment with intraventricular doses of VEGF. Both showed improved motor and memory functions compared with untreated mice.
Thau-Zuckman and colleagues subjected mice to traumatic brain/closed head injury and then infused VEGF into the lateral ventricles. In comparison with untreated mice, they found increased numbers of “proliferating cells in the subventricular zone and in the perilesion cortex,” indicating augmented angiogenesis at the site of injury and reduced lesion volumes there.
In summary, numerous investigators have shown in brain-injured animals that VEGF administered into the ventricle contiguous to the injury reduces the ensuing cerebral deficit for a time. Also VEGF injections in transgenic AD mice induced a transient angiogenesis and increased microvasculature. In her 2011 review on AD, Paula Grammas discussed many of the above reports and speculated about an “unexplored connections between angiogenesis and AD.” She inferred that this area may offer “new therapeutic approaches [so] desperately needed.” Also in 2011, Peter Carmeliet and Rakesh Jain wrote, “The revascularization of ischaemic tissues would benefit millions, but therapeutic angiogenesis remains an unmet medical need.”
The experimental brain injuries described above (carotid artery occlusion and local brain trauma) were acute cerebral events, in contrast with the slow development of senile dementias in people. Nevertheless, these animal studies suggest that providing angiogenic factors chronically to the human brain might retard or ameliorate cerebral microvasculopathies associated with aging.
Recent investigators have cautioned against the use of a single endothelial growth factor in therapeutic efforts to correct various vasculopathies, including strokes. As early as 2000 Yancopoulos et al. wrote that many members of the VEGF, angiopoietin and ephrin families interact in a complementary and coordinated manner to form functional vessels without leaks. (Recall that VEGF was originally called the vascular permeability factor.) In 2002 both Thurston and Visconti et al. reported on the synergism of VEGF and angiopoietin-1 in angiogenesis and warned that using VEGF alone risks the growth of leaky vessels. Similar reservations were expressed by Manoonkitiwongsa and colleagues for treating stroke victims and by Thau-Zuchman for traumatic brain injuries. Because several factors “in a cocktail” may be necessary to obtain functional capillaries (new or repaired), the plural term “factors” or “VEGF et al.” will be used in the discussion below, which describes six potential routes for administering angiogenic factors to the brain. They include the blood stream, the brain surface, the CSF (subarachnoid space), the brain parenchyma or the olfactory/trigeminal nerves.
VEGF et al. could be given intravenously over many months via a programmable minipump placed subcutaneously in the body. But parenteral treatment has the potential to stimulate angiogenesis throughout the body (possibly an untoward effect), rather than being limited to the brain. The desired localization might be achieved by linking the factors to an antibody specific for surface antigens on capillary endothelial cells—for example, transferrin receptors on the luminal surface of capillaries. This approach is suggested by the work of Friden and team, who coupled nerve growth factor (NGF) to OX-26, which is a mouse monoclonal antibody specific for transferrin receptors. NGF was detected within the rat brain capillaries and brain parenchyma when the complex was injected intravenously but not when NGF alone was tested.
Another parenteral approach might involve the angiogenic factors coupled to magnetic nanoparticles (MNP). Such a delivery system consists of three parts: a magnetic core (usually magnetite, Fe3O4), a surface coating (for example, polyethylene glycol) and a functional outer coating (the factors). When introduced into the circulation (or CSF), this MNP agent could be directed to the cerebral capillaries (or cerebral pia mater) and held there by a magnetic field over the skullcap. As Kumar and colleagues put in a Nanomedicine article,“ A simple external magnetic field is all that is necessary to target a drug [-coated ferro-nanoparticle] to a specific site inside the body,” and the drug can readily be retained there. For example, a study on stent angioplasty employed an agent to prevent stenosis (paclitaxel) being loaded onto magnetic nanoparticles and then subjected to a uniform induced magnetic field at the stent site. The authors reported a 4- to 10-fold higher concentration there of drug-loaded particles than of the agent injected alone.
The surface of the cerebral cortex is accessible via burr holes, a relatively simple procedure, now performed in an outpatient setting in 20 minutes or so and requiring only a local anesthetic to deaden pain in the epicranium. Angiogenic factors could be introduced by this route when incorporated in sustained release polymers of the type originally used by Folkman or in biodegradable polyanhydride wafers (Gliadel wafers). The latter were used by Attenello et al. and McGirt et al. to deliver anticancer agents directly to the site of excised gliomas in people. Similar studies in rats have employed four different intracranial resorbable, slow sustained release systems—surgical foam, a thermal gel depot, a microcapsule or biodegradable polymer beads. Such a slow-release device containing angiogenic factors could be placed on the pia mater covering the cerebral cortex and tested in persons with senile dementia in long term studies.
The spinal fluid circulates from the ventricles through the subarachnoid spaces over the pia mater covering the cerebral hemispheres. Introducing VEGF et al. by a slow pump into a ventricle would allow the factor also to reach the surface of the cerebral cortex. Parkinsonian patients have received nerve growth factor by infusion into the lateral ventricle over a 12-month period. A more direct route would entail injecting the factors into the subarachnoid space over the cerebral hemispheres through catheters (1 millimeter outer diameters) containing multiple draining holes—like those used in intraputamenal infusion studies.

Wikipedia
A sixth potential route involves the nasal cavity, where macromolecules may be absorbed across the olfactory membrane (olfactory receptor cells) and reach the brain parenchyma. For example, when gelfoam was soaked in a wheat germ agglutinin-horseradish peroxidase solution (WGA-HRP) and inserted into the nose of rats, neurons labeled with WGA-HRP were found “in the midbrain and pons and throughout the entire expanse of the olfactory cortex to the caudal pole of the cerebral hemisphere” according to M. T. Shipley. Similar studies were performed by Illum and X-Q Chen and colleagues, who used enzyme-linked immunosorbent assay to measure recombinant human nerve growth factor given by nose drops in the brains of anesthetized rats and found that concentrations reached 3,400 picomoles in the olfactory bulb within an hour and 660-1,300 picomoles in four coronal sections of the cerebral hemisphere taken from anterior to posterior. They suggested in their 1989 paper using this route for the long term treatment of Alzheimer’s disease with NGF. Perhaps in the distant future people will regularly “take a pinch of snuff” containing appropriate angiogenesis factors to delay/relieve senile dementias. Eyedrops should also be studied as a route for introducing agents into the brain.
The NAG hypothesis has two constructs: First, during aging there is a decline in capillary density due to waning neuroangiogenesis, which varies in different persons as determined by their genetic background. Second, this normal age-related decline may be accelerated by environmental factors depressing the usual levels of neuroangiogenic factors or may possibly be delayed by administration of exogenous angiogenic factors directly to the brain.
This hypothesis, if valid, has practical implications for preventing, reducing or possibly ameliorating Alzheimer’s disease and other senile dementias. Six therapeutic approaches have been discussed. In any future clinical studies, investigators will be challenged to determine the most effective and safest combination of neuroangiogenic factors to preserve or revive a healthy cerebral microcirculation.
On a lighter note, the mildest examples of a defective cerebral microcirculation may be those experienced by adults during casual conversations (“senior moments”) and by students during exam times, when perhaps several capillaries may be transiently compromised. Here I’m reminded of an old Sinatra song entitled “High Hopes,” about an ant who sought to topple a rubber tree plant. One chorus ends with “Oops, there goes another rubber tree plant.” When I can’t recall a familiar name or word, I now think to myself, “Oops, there goes another capillary in my brain.”
I must acknowledge here the countless investigators in this field whose contributions could not be cited in this short essay but provide silent support for the NAG hypothesis. I am greatly indebted to the Medical Center Library of the University of Kentucky and its expeditious staff member, Mrs. Amanda Williams. Finally, I acknowledge the long term support of the late Ch. Tray.
Click "American Scientist" to access home page

American Scientist Comments and Discussion
To discuss our articles or comment on them, please share them and tag American Scientist on social media platforms. Here are links to our profiles on Twitter, Facebook, and LinkedIn.
If we re-share your post, we will moderate comments/discussion following our comments policy.